Introduction
Causes
Demographics
Signs and symptoms
Diagnosis
Treatment
Prognosis
Dermatofibrosarcoma protuberans is an uncommon skin tumour arising in the deeper layer of the skin (the dermis). It grows slowly but has a tendency to recur after excision. Luckily, it rarely spreads to other sites beyond the skin.
The cause of dermatofibrosarcoma protuberans is unknown, but an injury to the affected skin may be a predisposing factor. Recent advances show tumour cells carry abnormal chromosomes within the tumour cells—t(17;22)(q22;q13)—resulting in the fusion gene COL1A1-PDGFB. This encodes a protein that causes a tumour to grow by autocrine overproduction of platelet-derived growth factor (PDGF).
Dermatofibrosarcoma protuberans is rare and affects less than 1 person in every 100,000 inhabitants per year.
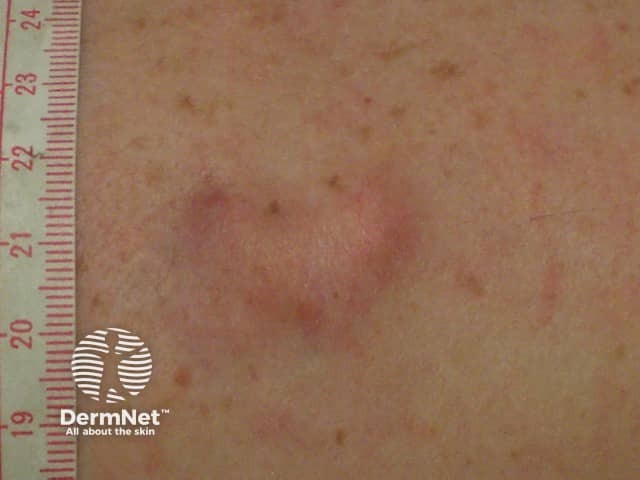
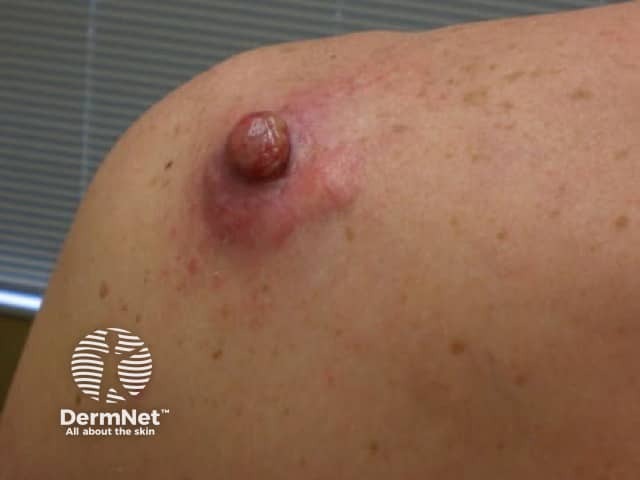
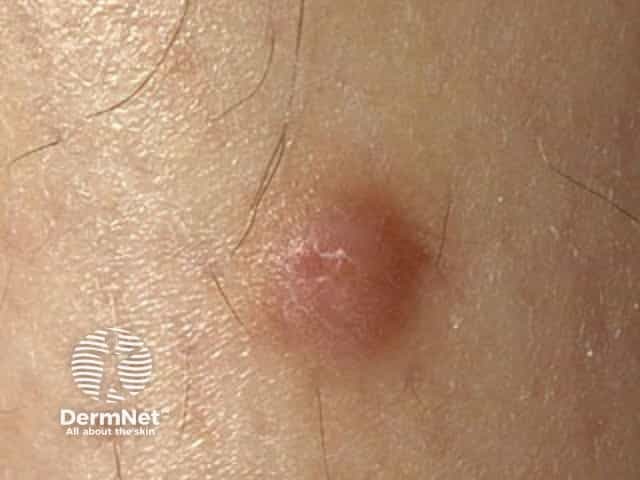
Dermatofibrosarcoma protuberans usually presents as a painless thickened area of skin (plaque) and/or nodule that feels rubbery or firm to touch and is fixed to the underlying skin. It may be red-brown or skin coloured. It usually grows very slowly over months to years.
Rarely it presents as a soft depressed area of the skin making the diagnosis even more difficult. Dermatofibrosarcoma protuberans may range in size from 0.5–25 cm in diameter. Fifty to sixty per cent of tumours arise on the trunk, often in the shoulder and chest area. The remaining 35% of tumours are found on the limbs and 10 to 15% on the head and neck region.
Dermatofibrosarcoma protuberans is often diagnosed when it enters a more rapid growth phase giving rise to larger lesions. Neglected tumours may reach large proportions.
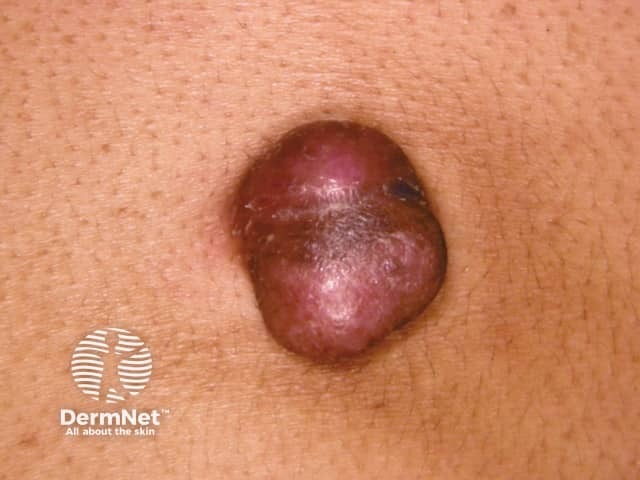
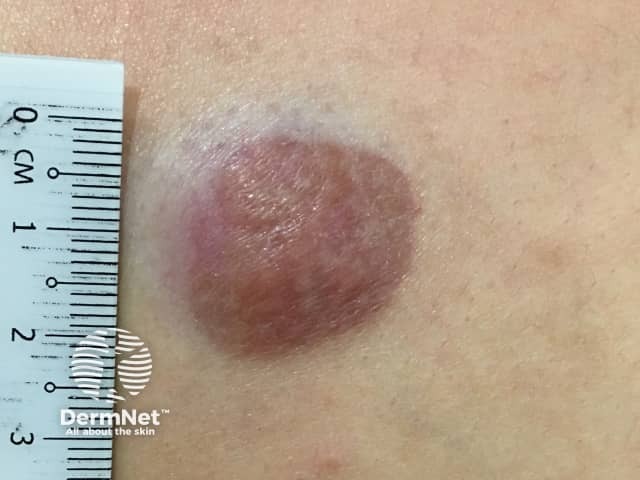
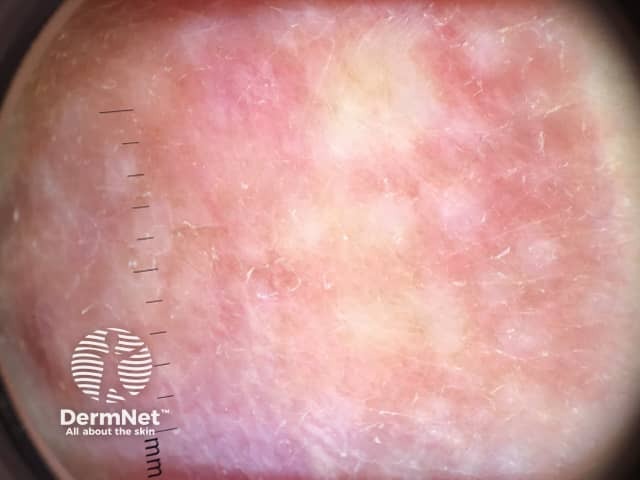
The absence of symptoms often leads to a delay in diagnosis of dermatofibrosarcoma protuberans. Redness and pain only occur in 15% of cases. It is often mistaken for other skin conditions, particularly in the early stages. Dermoscopy is not diagnostic as the features of dermatofibrosarcoma protuberans are nonspecific. Skin biopsy, including the deep dermis and subcutis, is needed to confirm the diagnosis. The abnormal cells are positive for CD34.
Dermatofibrosarcoma protuberans has a characteristic appearance under the microscope with densely arranged spindle-shaped cells. It may be difficult to assess complete removal due to extensions widely in the skin and deeper structures. It is important to identify fibrosarcomatous dermatofibrosarcoma protuberans, a more aggressive tumour, which requires more aggressive treatment. This shows a high mitotic rate particularly deep within the tumour.
Dermatofibrosarcoma protuberans's chromosomal abnormalities can be detected using reverse transcription-polymerase chain reaction (RT-PCR) or fluorescence in-situ hybridisation (FISH).
In most cases of dermatofibrosarcoma protuberans, no other investigations are necessary. If there is suspicion of metastasis or there is fibrosarcomatous transformation, lymph node ultrasound, chest X-ray and pelvic ultrasound scan may be arranged.
Treatment of dermatofibrosarcoma protuberans and dermatofibrosarcoma protuberans with fibrosarcomatous transformation consists of wide excision of the lesion including deep fascia, with 1–3 cm margin of normal skin. It may take more than one surgical procedure to ensure complete removal of a tumour.
Mohs micrographic surgery, which is a special surgical technique to control tumour margins, is sometimes used to check that all the abnormal cells have been excised. Recurrence after Mohs surgery is reported to be around 1%.
Radiotherapy is sometimes used in addition to surgery if a tumour cannot be completely removed by surgery. Chemotherapy is ineffective.
The tyrosine kinase inhibitor imatinib mesylate is used to treat rare cases of local advanced inoperable or metastatic dermatofibrosarcoma characterised by COL1A1-PDGFB fusion gene, with 50% response rates.
Follow-up with clinical examination of the site of the dermatofibrosarcoma protuberans is recommended every 6 months for 5 years, and then annually.
The tumour only metastasises in 5% of cases. It spreads in 1% via lymphatic vessels to the regional lymph glands and in 4% via the bloodstream, most commonly to the lung followed by the brain, bone and heart.
Local recurrences arise in 11–20% of cases, usually within 3 years of initial surgery, so follow-up is important. The recurrences are treated surgically as described for the original primary tumour.