What is autoimmune polyglandular syndrome type 1?
Autoimmune polyglandular syndrome type 1 (APS1) is an autoimmune condition that results in insufficiencies of multiple endocrine glands. It is also known as autoimmune polyendocrine syndrome type 1, polyendocrinopathy-candidiasis–ectodermal dystrophy (APECED), Whitaker syndrome, and candidiasis-hypoparathyroidism–Addison disease syndrome, among its many other names.
APS1 was first described by Dr Thomas Addison in the 19th century.
Who gets autoimmune polyglandular syndrome type 1?
APS1 is inherited, with women and girls being slightly more likely than men and boys to develop the syndrome. It most often occurs in particular ethnic populations due to consanguinity or the clustering of descendants from a common family founder. It is most prevalent in:
- Iranian Jews (1 in 9000)
- Sardinians (1 in 14,400)
- Finns (1 in 25,000).
APS1 is rare in other populations.
What causes autoimmune polyglandular syndrome type 1?
APS1 is caused by gene mutations in the autoimmune regulator gene, AIRE, on chromosome 21q22. It is inherited in an autosomal recessive pattern (two copies of an abnormal gene must be present for the syndrome to develop). These gene mutations lead to autoantibodies and cause chronic inflammatory cell infiltrates in the affected organs.
What are the clinical features of autoimmune polyglandular syndrome type 1?
Symptoms most often appear in children aged 3–5 years, most cases of APS1 have appeared by early adolescence, and all cases by the time an individual is in their early 30s.
APS1 is based on three main clinical features:
- Mucocutaneous candidiasis affecting the skin and mucous membranes
- Hypoparathyroidism, resulting in numbness and tingling in the face and limbs, muscle cramps and aches, weakness and fatigue due to low levels of circulating calcium
- Addison disease, an insufficiency of the adrenal glands, presenting with changes in skin pigmentation, loss of appetite and weight loss, fatigue, low blood pressure and fatigue.
While less common, other possible features of this syndrome can include:
-
- Hypogonadotropic hypogonadism
- Pernicious anaemia
- Chronic active hepatitis
- Asplenia
- Keratoconjunctivitis
- Interstitial nephritis
- Diabetes mellitus type 1
- Cholelithiasis
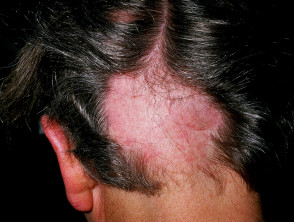
- Alopecia areata
- Malabsorption
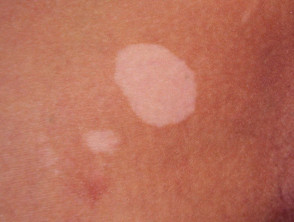
- Vitiligo.
Skin signs of APS type 1
How is autoimmune polyglandular syndrome type 1 diagnosed?
If an individual presents with evidence of more than one endocrine deficiency, further testing can be done to confirm autoimmune polyglandular syndrome type 1 (APS1), including:
- A serum autoimmune screen
- End-organ function tests.
Additional possible blood tests can include testing of testosterone, oestradiol, follicle-stimulating hormone (FSH), luteinising hormone (LH), prolactin, adrenocorticotropic hormone (ACTH), plasma renin activity, electrolyte levels, and a complete blood count.
Skin swabs and scrapings may also be taken to detect Candida albicans.
How is autoimmune polyglandular syndrome type 1 treated?
The treatment of APS1 will depend upon its specific features.
- Mucocutaneous candidiasis is treated with oral antifungal agents, such as fluconazole or itraconazole.
- Hypoparathyroidism is treated with a combination of oral calcium and vitamin D (usually, calcitriol).
- Addison disease is treated with oral corticosteroids and mineralosteroids. Adrenal gland transplants may also be indicated.
What is the outcome for autoimmune polyglandular syndrome type 1?
The prognosis for APS1 is variable. Survival rates have improved greatly since the 1970s.