What is autoimmune polyglandular syndrome type 2?
Autoimmune polyglandular syndrome type 2 (APS2) is the most common of the immunoendocrinopathy syndromes.
- It is characterised by the involvement of two or more organs.
- It usually presents as a combination of Addison disease with either thyroid disease or diabetes mellitus type 1.
APS2 is also known as Schmidt syndrome.
Who gets autoimmune polyglandular syndrome type 2?
APS2 is inherited in an autosomal dominant pattern; that is, an abnormal gene from one parent can cause disease and this happens even when the matching gene from the other parent is normal. A parent with an autosomal dominant condition has a 50% chance of having a child with the condition.
- APS2 is rare — it affects 14 to 20 people out of every million.
- It is 3–4 times more common in women than in men.
What causes autoimmune polyglandular syndrome type 2?
Researchers theorise that APS2 develops when a person with genetic susceptibilities is exposed to an autoimmune trigger. Initially, autoimmune activity is asymptomatic. Progressive organ damage and inflammatory infiltration occur over time.
What are the clinical signs of autoimmune polyglandular syndrome type 2?
The clinical signs of APS2 include the signs of Addison disease, autoimmune thyroid disease and/or diabetes mellitus type 1 and most commonly appear in people aged 20–30 years.
The signs and symptoms of APS2 vary depending on the specific clinical manifestations seen in an individual; they may include:
-
- Diabetes mellitus type 1 that can present with thirst, hunger, excessive urine production, weight loss and fatigue
- Graves disease characterised by palpitations, intolerance to heat, decreased energy levels, anxiety, infrequent menstruation, weight loss and muscle weakness.
- Hashimoto thyroiditis that can present with memory problems, myalgia, fatigue, weakness sleepiness, constipation and cold intolerance
- Addison disease causing nausea and vomiting, loss of appetite, weight loss and fatigue
- Pernicious anaemia that may present with pallor and tiredness
- Coeliac disease presenting with abdominal pain and cramping, fatty bowel motions, and weight loss.
Skin signs of APS type 2
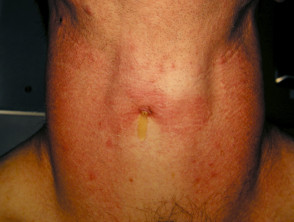
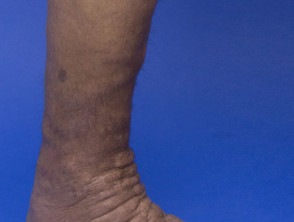
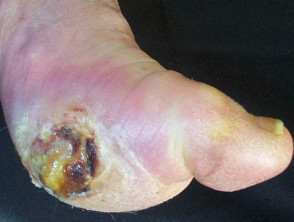
How is autoimmune polyglandular syndrome type 2 diagnosed?
APS2 is diagnosed in an individual who has multiple autoimmune diseases. Tests used to diagnose APS2 may include:
- Screening for type 1 diabetes, autoimmune thyroid conditions and coeliac disease
- Serum autoimmunity tests
- End-organ function tests.
Other tests may include gonadotropin levels, thyroid tests (thyroid-stimulating hormone [TSH], triiodothyronine [T3] and thyroxine [T4]), plasma renin activity, serum electrolytes, fasting blood glucose and a complete blood count.
How is autoimmune polyglandular syndrome type 2 treated?
The treatment for APS2 depends upon which organs are affected, and may include:
- Lifelong insulin treatment for type 1 diabetes
- Thyroid replacement therapy for Hashimoto thyroiditis
- Intramuscular injections of vitamin B12 for pernicious anaemia
- Corticosteroid replacement for Addison disease
- Antithyroid medications, radioactive iodine treatment, or thyroidectomy for Graves disease
- Gluten-free diet for coeliac disease
- Immunosuppressant medications.
What is the outlook for autoimmune polyglandular syndrome type 2?
The outlook for APS2 is variable and depends upon which specific organs or glands become affected by the syndrome.