What is Cronkhite–Canada syndrome?
Cronkhite–Canada syndrome is a rare, sporadic disorder characterised by generalised gastrointestinal polyposis and dermatological manifestations consisting of hyperpigmentation, hair loss, and nail dystrophy.
Cronkhite–Canada syndrome was first described by Leonard Cronkhite and Wilma Canada in 1955.
Clinical features of Cronkhite-Canada-syndrome
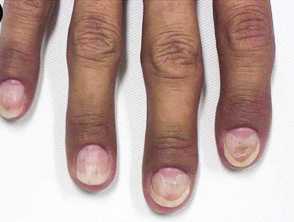
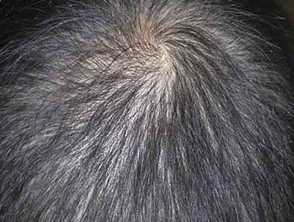
Images from: Onozato Y, Sasaki Y, Abe Y, et al. Cronkhite-Canada syndrome associated with gastric outlet obstruction and membranous nephropathy: a case report and review of the literature. Intern Med. 2020;59(22):2871-7.
Who gets Cronkhite–Canada syndrome?
- Over 500 reported cases of Cronkhite–Canada syndrome in the medical literature
- 75% of reported cases are Japanese
- Sporadic with no family history
- Male predominance 2-3:1
- Average age of onset is around 60 years.
What causes Cronkhite–Canada syndrome?
The cause of Cronkhite–Canada syndrome is unknown. Physical and emotional stress are commonly reported triggers.
Autoimmune diseases reported in association with Cronkhite-Canada syndrome include vitiligo, Hashimoto thyroiditis, systemic lupus erythematosus, rheumatoid arthritis, and systemic sclerosis. The ANA is often positive.
The cutaneous manifestations may be due to nutritional deficiencies caused by the gastrointestinal changes.
What are the clinical features of Cronkhite–Canada syndrome?
Common presenting symptoms in one large case series were:
- Nail dystrophy (93%)
- Diarrhoea (90%)
- Hair loss (87%)
- Hyperpigmentation (87%)
- Impaired or reduced sense of taste (60%)
Mucocutaneous features
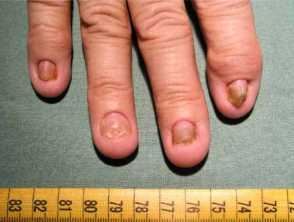
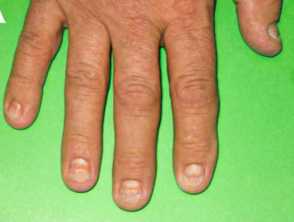
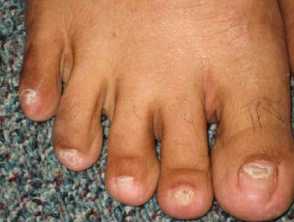
- Nail dystrophy - including onycholysis, koilonychia, onychomadesis
- Hair loss - may be patchy or diffuse, mild to severe, from the scalp, eyebrows, eyelashes, beard
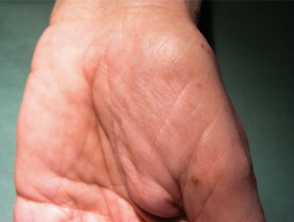
- Hyperpigmentation - diffuse or well-defined brown macules on the palms and soles, upper limbs, face, and chest
- Swelling and loss of papillae on the tongue described with hypogeusia
Onychodystrophy in Cronkhite-Canada syndrome
Palmar pigmentation of Cronkhite-Canada syndrome
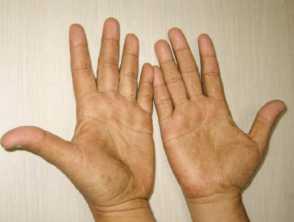
Figures 1&4 from: Kopáčová M, Urban O, Cyrany J, et al. Cronkhite-Canada syndrome: review of the literature. Gastroenterol Res Pract. 2013;2013:856873.
Figure 2 from: Schulte S, Kütting F, Mertens J, et al. Case report of patient with a Cronkhite-Canada syndrome: sustained remission after treatment with corticosteroids and mesalazine. BMC Gastroenterol. 2019;19(1):36.
Figures 3&5 from: Kao KT, Patel JK, Pampati V. Cronkhite-Canada syndrome: a case report and review of literature. Gastroenterol Res Pract. 2009;2009:619378.
Gastrointestinal features
- Nausea and vomiting, loss of appetite, and weight loss, often with cachexia
- Upper abdominal pain, watery diarrhoea, and melaena
- Lactose intolerance
The formation of polyps throughout the gastrointestinal tract (but sparing the oesophagus) is the diagnostic finding on endoscopy.
What are the complications of Cronkhite-Canada syndrome?
- Anaemia
- Protein-losing enteropathy causing hypoproteinaemia and peripheral oedema
- Malnutrition including vitamin deficiencies
- Intussusception and rectal prolapse
- Pancreatitis
- Gastrointestinal malignancy - gastric and colorectal - 15%
- Portal vein thrombosis
- Membranous glomerulonephritis
How is Cronkhite–Canada syndrome diagnosed?
Cronkhite-Canada syndrome should be considered in a patient presenting with the typical gastrointestinal symptoms and skin signs, and confirmed on endoscopy with characteristic histology of dilated mucosal glands, oedematous stroma, and inflammatory infiltrate.
Blood tests will often show anaemia, hypoproteinamia, electrolyte disturbances, nutritional deficiencies, and positive ANA.
Faecal occult blood is positive.
What is the differential diagnosis of Cronkhite-Canada syndrome?
- Familial polyposis
- Peutz-Jeghers syndrome
- Cowden disease
How is Cronkhite–Canada syndrome treated?
- Nutritional support
- Systemic corticosteroids are the mainstay of treatment
- Other immune modulating agents if steroid-resistant or steroid-dependent
What is the outlook for Cronkhite–Canada syndrome?
The outlook for Cronkhite–Canada syndrome was initially poor, with mortality rates in the first 5 years after diagnosis of around 50%. Early aggressive treatment with systemic steroids has improved survival and results in the reversal of symptoms and signs in 2-4 months. Steroid dose should be reduced slowly as rebound flare is common if dose reduction is too rapid.