Introduction
Dowling-Degos disease is an autosomal dominant skin disorder characterised by progressive pigmentation of the skin within body folds. Association with loss-of-function mutations in KRT5 (encoding keratin 5) is documented.
Histology of Dowling-Degos disease
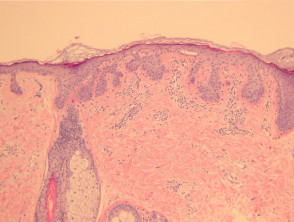
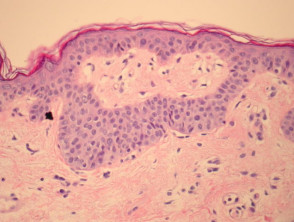
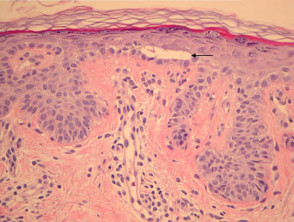
Epidermal changes in Dowling-Degos disease include hyperkeratosis, often with small horn cysts and thinning of the suprapapillary epidermis. Thin epithelial strands extend into the superficial dermis from the epidermis and hair follicles resulting in an ‘antler-like’ pattern (Figures 1, 2). Epidermal hyperpigmentation is usual although rare non-pigmented variants are reported. The hair follicle infundibulum may be dilated. Dermal perivascular lymphohistiocytic infiltrate (figures 1,2,3) or a lichenoid tissue reaction pattern may be seen.
Dowling-Degos disease pathology
Differential diagnosis of Dowling-Degos disease
Galli-Galli disease is a subtype of Dowling-Degos disease and is distinguished by the additional finding of non-dyskeratotic acantholysis (Figure 3, arrow). Other clinical and histological features are identical to Dowling-Degos disease.
Seborrhoeic keratosis — in Dowling-Degos disease, epithelial strands extend from the epidermis and hair follicles. The clinical presentation is very helpful in differentiating these conditions.
Haber syndrome — shared histological features with Dowling-Degos disease; however, the clinical presentation is distinct with facial rosacea-like lesions, pitted scars and seborrhoeic keratosis-like lesions in the trunk and flexures. Haber syndrome is not associated with keratin 5 mutations.