What is Gardner syndrome?
Gardner syndrome is a variant of familial adenomatous polyposis (FAP) that is associated with extra-colonic features. It is an inherited disease that is characterised by gastrointestinal polyps, multiple osteomas (benign bone tumours), and various skin and soft tissue tumours.
Polyps tend to form at puberty with the average age of diagnosis around 25 years of age. Polyps have an almost 100% risk of undergoing malignant transformation resulting in colorectal cancer, therefore timely detection of Gardner syndrome is essential.
Who gets Gardner syndrome?
The incidence of familial adenomatous polyposis (FAP) is 1 case in 7500 live births. It is due to a congenital autosomal dominant mutation in around 80% of patients. The remaining 20% are spontaneous mutations, with no associated family history.
Although colonic polyps begin to form in puberty, the average age at which Gardner syndrome is diagnosed is 22 years. Osteoma formation usually precedes polyposis. Progression to malignancy is generally observed at age 30–50 years. The average age by which malignancy is detected in patients with Gardner syndrome is 39 years.
What causes Gardner syndrome?
Gardner Syndrome is due to mutations on the adenomatous polyposis coli (APC) gene on chromosome 5q22. The gene plays a role in tumour suppression. Gardner syndrome is inherited as an autosomal dominant trait, so an affected person has a 50% chance of passing on the gene to each of their children.
Genetics of Gardner syndrome*
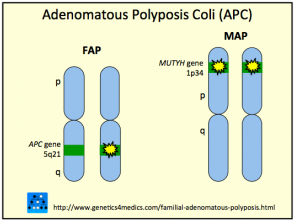
*Image courtesy of Genetics 4 Medics
What are the clinical features of Gardner syndrome?
Clinical features of Gardner syndrome are both cutaneous and non-cutaneous.
Cutaneous features
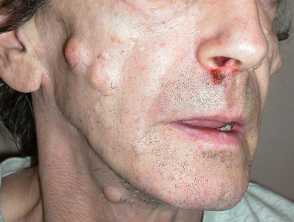
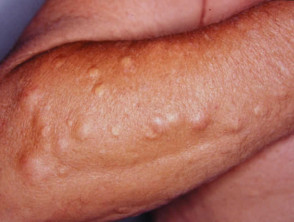
The most noticeable cutaneous feature is the appearance of epidermoid cysts. These can be differentiated from ordinary epidermoid cysts by the following factors:
- Epidermoid inclusion cysts of Gardner syndrome (50–65%) often occur at an earlier age (around puberty) than ordinary cysts
- Epidermoid cysts occur in less common locations such as the scalp, trunk, and limbs compared to ordinary cysts
- Cysts tend to be multiple in over half of the patients with Gardner syndrome
- As with ordinary epidermoid cysts, cysts in Gardner syndrome are usually asymptomatic, however in some cases, they may be pruritic (itchy) or inflamed, and they may rupture causing local inflammation
- Sometimes the cysts have hybrid features with pilomatricoma-like histopathology.
Other cutaneous features include fibromas, lipomas, leiomyomas, and neurofibromas.
Non-cutaneous features
- Gastrointestinal polyps that nearly always transform into colonic adenocarcinomas (colon cancer). The polyposis predominantly affects the left colon (80–90% of cases). Patients may be asymptomatic or report symptoms such as abdominal pain, change in bowel habit, or blood in stool.
- Desmoid tumours — deep fibromatoses (eg, in the abdominal wall) that occur more commonly in patients with familial adenomatous polyposis (FAP) than in the general population; benign but often locally aggressive.
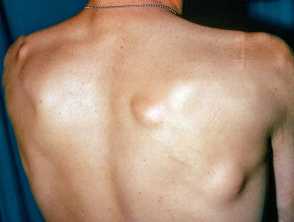
- Osteomas — these benign bone tumours are essential in making the diagnosis of Gardner syndrome. They occur most commonly in the mandible (jawbone) but may also grow in the skull and long bones. Osteoma formation often precedes polyposis.
- Dental abnormalities — as well as osteomas in the jaw there may be other dental abnormalities such as unerupted teeth, supernumerary teeth, and caries.
- Congenital hypertrophy of the retinal pigment epithelium (CHRPE) — multifocal pigmented lesions of the fundus in the eye, seen in 80% of patients. These lesions may be present shortly after birth and can be the first marker of the disease.
- Patients with colorectal cancer and FAP appear to be more at risk for other cancers, including the cribriform variant of papillary thyroid cancer. In women younger than 35 years, the risk is 160 times greater than normal. Other malignant tumours include periampullary adenocarcinoma, hepatoblastoma, and multifocal cholangiocarcinomas.
What are the complications of Gardner syndrome?
- Colorectal cancer (extremely high risk; almost certain by age 40).
- Risk of other FAP-related malignancies (eg, gastrointestinal, liver, adrenal, or thyroid cancer).
- Dental abnormalities that may require dental procedures.
- Cosmetic or functional concerns from cysts, other cutaneous features, or osteomas.
- Desmoid tumours are benign but can be locally aggressive and over time damage or compress other structures.
How is Gardner syndrome diagnosed?
Gardner syndrome is diagnosed based on the following features:
- ≥100 colorectal polyps OR ≤100 polyps and a family member with familial adenomatous polyposis (FAP) or Gardner syndrome
- Osteomas
- Soft tissue tumours such as epidermoid cysts, fibromas, and desmoid tumours
- Adenomatous polyposis coli (APC) gene mutation.
Radiological studies are essential for patients and family members with suspected Gardner syndrome.
- Images of the long bone may show osteomas.
- Images of the mandible at an early age may show subtle defects.
- Eye exams at an early age can detect pigmented lesions of the fundus.
- Colonoscopy and other invasive tests to check for polyp involvement every 1–2 years.
What is the differential diagnosis for Gardner syndrome?
Differential diagnoses for some features of Gardner syndrome include:
- Ordinary epidermoid cysts
- Other forms of cutaneous cysts and pseudocysts
- Classic familial adenomatous polyposis (FAP)
- Attenuated FAP
- Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS)
- Turcot syndrome
- Hereditary nonpolyposis colorectal cancer (Lynch syndrome)
- MUTYH-associated polyposis (MAP)
- Peutz-Jeghers syndrome
- Cronkhite-Canada disease
- Juvenile polyposis syndrome (JPS)
- Multiple endocrine neoplasia type 2B.
What is the treatment for Gardner syndrome?
- Epidermoid cysts: treatment is similar to that of ordinary cysts and involves excision. Occasionally, intralesional steroid injections may be used if the cysts are inflamed.
- Desmoid tumours: management options may include monitoring (wait-and-see), surgical removal, or (eg, in the case of unresectable tumours) systemic therapies such as non-steroidal anti-inflammatories or chemotherapy.
- Osteomas: may also require excision, if a nuisance or severely deforming.
- Regular lower gastrointestinal endoscopy surveillance: recommended to monitor polyps and screen for malignancy. Over 20 polyps may be an indication for surgical removal of the colon (colectomy) due to the high risk of polyps developing into cancer.
- Regular gastroduodenoscopy: every 1–2 years is also recommended.
- Annual physical examination: including skin check and palpation of the thyroid is recommended.
- Thyroid function evaluations: may be required beginning in the late teenage years.
What is the outcome of Gardner syndrome?
There is no cure for Gardner syndrome. A person with an APC gene mutation will develop colon cancer with almost 100% certainty by around age 40 if surgery is not performed and polyp growth controlled.