What is BRCA1-associated protein-1?
BRCA1-associated protein-1 (BAP1) is a nuclear protein encoded by the BAP1 tumour-suppressor gene located on chromosome 3 (locus 3p21.1). The BAP1 protein acts as a deubiquitinating enzyme — removing ubiquitin, a regulator of the degradation of proteins — and is involved in many key cellular functions including:
- DNA repair
- Deubiquitination of histones (proteins that 'package' DNA into chromosomes)
- Chromatin remodelling (the process that opens up the coils of DNA to allow gene expression)
- Transcription (the process that copies DNA into RNA to make proteins)
- Cell-cycle regulation [1].
For more information about genes associated with melanoma, see the DermNet pages on Genes and melanoma, Genetics of melanoma, and Genetic testing for melanoma.
Who gets the germline BAP1 mutation?
The prevalence of germline BAP1 mutations in the general population is not known. At least 215 affected individuals from 87 families have been reported in the literature to date (as of September 2018) [2].
What causes germline BAP1 mutation?
Germline BAP1 mutations are inherited in an autosomal dominant pattern, meaning that only one parent has to carry the mutated gene for their child to inherit the condition. Each somatic cell of the affected offspring carries one mutant BAP1 allele and a normal wild-type allele. An additional somatic mutation must be acquired in the wild-type allele for the cell to lose BAP1 tumour-suppressor function.
The exact genotype–phenotype correlation between the location and type of mutation and cancer risk is not known [3]. Some mechanisms of mutagenesis have been reported, the majority of which result in a truncated protein [4].
Not all mutations are damaging to protein function. Significant BAP1 mutations are those that affect either the nuclear localisation sequence, resulting in retention of the protein in the cytoplasm, or the ubiquitin carboxy-terminal hydrolase catalytic domain, affecting deubiquitinating activity [2,5].
What conditions are associated with the germline BAP1 mutation?
In 2011, an inherited cancer predisposition syndrome was proposed in association with the germline BAP1 mutation [6–8]. Carriers of the mutation have a high frequency of four main malignancies; these being:
- Malignant mesothelioma (pleural and peritoneal)
- Uveal (ocular) melanoma
- Cutaneous melanoma
- Renal cell carcinoma.
A wide range of other tumours have been reported in association with the mutation, including a higher incidence of:
- Basal cell carcinoma
- Breast cancer
- Cholangiocarcinoma
- Meningioma
- Neuroendocrine tumours
- Thyroid cancer.
Although it has a BRCA1 protein-binding site, mutations in the BAP1 gene are not common in BRCA1-associated breast cancer. Further studies are needed for confirmation.
The lifetime risk of developing cancer in those with a harmful germline mutation is not known. Large population studies are required to estimate the true incidence of cancer in those with the mutation. In the 215 cases reported, 60 individuals (28%) have developed uveal melanoma, 48 mesotheliomas (22%), 38 (18%) cutaneous melanoma, and 20 (9%) renal cell carcinoma [2]. However, germline BAP1 mutations account for only a small proportion of these cancers overall; approximately 1.6% [8,9] to 4% [10,11] of uveal melanomas, 1–2% [12] of malignant mesothelioma, 0.01% [13] to 0.63% [6,10] of cutaneous melanoma, and 1.2% [14] of renal cell carcinomas.
Cancer in BAP1 mutation carriers is reported in a younger median age of onset and has a varying prognostic significance compared with the general population (table 1). Both somatic and germline BAP1 loss is associated with a more aggressive phenotype in uveal melanoma [9,15,16] and renal cell carcinoma [17,18], possibly shorter disease-free survival and melanoma-specific survival in cutaneous melanoma [19], but a better overall survival in mesothelioma [20–22].
| Cancer Type | Median age of onset in germline BAP1 mutant (range) [3] | Median age of onset (general population) [3] | Frequency (%) in a cohort of 215 germline BAP1 mutation positive patients [2] | Prognosis of germline BAP1 associated tumours |
|---|---|---|---|---|
| Uveal melanoma | 51 (16–72) | 62 | 28 | Worse |
| Malignant mesothelioma | 53 (34–85) | 74 | 22 | Better |
| Cutaneous melanoma | 45 (21 or 25–72) | 58 | 18 | Possibly worse |
| Renal cell carcinoma | 47 (36–72) | 64 | 9 | Worse |
What is a BAP1-inactivated melanocytic tumour?
The BAP1-inactivated melanocytic tumour is a rare kind of melanocytic naevus and is one of the earliest and most common clinical manifestations of the germline BAP1 mutation. The tumour is characterised by loss of BAP1 expression on immunoperoxidase staining; this was first described by Thomas Wiesner in 2011 [6]. BAP1-inactivated melanocytic tumours occur in approximately 67–75% of germline BAP1 carriers, typically after the second decade of life [2,5]. These tumours were initially histologically classified as atypical Spitz tumours because of the overlapping features between Spitz naevi and spitzoid melanoma, but at a molecular level, BAP1 loss and BRAF mutation are not seen in the majority of atypical Spitz tumours.
In the 2018 World Health Organization's WHO classification of skin tumours, BAP1-inactivated (4th Ed, 2018), the BAP1 inactivated melanocytic tumours are classified as BAP1-inactivated naevus and BAP1-inactivated melanocytoma (if there are atypical features) [23].
Other names include BAP-oma, Wiesner naevus, melanocytic BAP1-mutant atypical intradermal tumour, and naevoid melanoma-like melanocytic proliferation.
BAP1-inactivated melanocytic tumours may be sporadic in individuals without the BAP1 mutation or the germline BAP1 familial syndrome [6].
What are the clinical features of BAP1-inactivated melanocytic tumour?
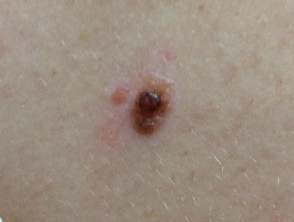
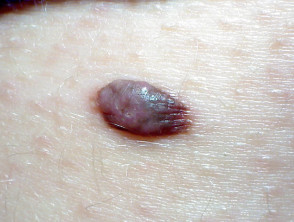
BAP1-inactivated melanocytic tumours appear from the second to third decade of life. They are circumscribed pink, orange, tan or skin-coloured papules, with an average size of 5 mm. Other features include:
- A dome shape or pedunculated morphology
- A clinical similarity to dermal naevi
- Five to 50 lesions per individual
- A typical location on the scalp and trunk.
BAP1-inactivated melanocytic tumour
What are the dermoscopic features of BAP1-inactivated melanocytic tumour?
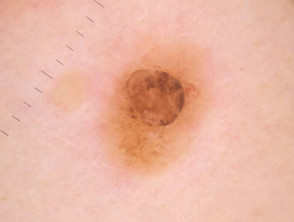
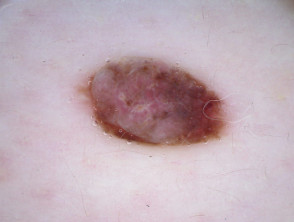
The dermoscopy of the BAP1-inactivated melanocytic tumour is not well described (case reports only).
- There may be a central pink, structureless area, with a varying distribution of peripheral pigmented globules.
- There may be polymorphous vessels centrally [2].
- Some lesions have a central asymmetrical multicomponent pattern with a peripheral reticular globular pattern, which may reflect the adjacent common naevus component from which the tumour may evolve [23,24].
- Other lesions have a structureless, whitish central area with irregular brownish-grey dots or blotches and peripheral irregular, linear vessels [24].
Dermoscopy of BAP1-inactivated melanocytic tumour
What are the histological features of BAP1-inactivated melanocytic tumour?
A BAP1-inactivated melanocytic tumour is predominantly based in the dermis (figure a), but it may be combined with a smaller adjacent junctional area that has more regular naevoid cells (which represents a common naevus component).
- BAP1-inactivated melanocytic tumours are composed mainly of large epithelioid (or spitzoid) melanocytes with copious eosinophilic cytoplasm and large vesicular nuclei (figure b, which also shows surrounding smaller regular melanocytes, and figure c).
- Spitzoid features include marked cytological atypia with nuclear pleomorphism, hyperchromatism, and predominant nucleoli — however, these tumours lack the Kamino bodies and spindle-shaped melanocytes, that are typically seen in Spitz naevi [25].
- To reflect the spectrum of atypia demonstrated by BAP1-inactivated cutaneous tumours, the WHO terminology classifies those with benign-appearing naevoid cells and minimal atypia as BAP1-inactivated naevi, and those with highly atypical large epithelioid cells and marked pleomorphism as BAP1 melanocytomas [23].
- There is low mitotic activity, and tumour-infiltrating lymphocytes are often present.
A BAP1-inactivated melanocytic tumour is BAP1 negative (biallelic loss) and has melanocytes. Note there is a loss of BAP1 expression on immunohistochemistry (IHC) of larger epithelioid melanocytes, while regular melanocytes retain BAP1 expression (figure d).
Figure 1. Histopathology of BAP1-inactivated melanocytic tumour
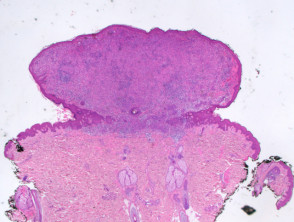
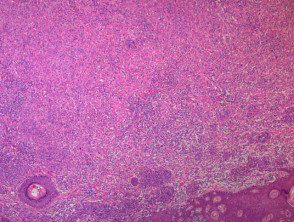
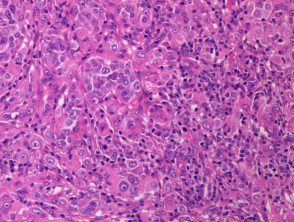
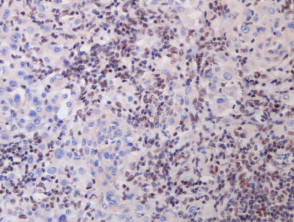
BAP1-deficient lesion. Male 14, back with a pedunculated lesion. Images supplied by Professor Richard Scolyer.
What is the malignant potential of the BAP1-inactivated melanocytic tumour?
There is very little known about the natural history of BAP1-inactivated melanocytic tumour or the phenotypic or histological features of primary cutaneous melanoma in this cohort. Despite high lesion counts in patients with BAP1-inactivated melanocytic tumours, there is a low rate of cutaneous melanoma [2]. Biallelic BAP1 loss and BRAF mutation is not sufficient for melanoma development [6]. There have been some reports of the transformation of a BAP1-inactivated melanocytic tumour to melanoma [26]. Depending on the degree of atypia, lesions may be classified as having uncertain malignant potential. They typically have an indolent course [27].
How is a BAP1-inactivated melanocytic tumour managed?
There are no prescriptive guidelines for the management of BAP1-inactivated melanocytic tumour. Any identified BAP1-inactivated melanocytic tumour should be closely followed with sequential digital dermoscopic imaging and excised if a change occurs. If they are excised, complete excision with histologically clear margins is mandatory. Consideration should be given to discussing lesions with uncertain malignant potential at a melanoma multidisciplinary meeting.
Who should be considered for genetic screening?
Indications for genetic testing include a strong personal or family history of the four main malignancies, coupled with multiple melanocytic naevi resembling BAP1-inactivated melanocytic tumours or atypical epithelioid tumours with spitzoid features on pathology [3,27].
Patients should be referred for genetic counselling before the decision to test for the genetic mutation. When referred for testing, the clinician should keep in mind that currently there are no formalised cancer surveillance plans for patients diagnosed as positive for the germline BAP1 mutation.
How is the germline BAP1 mutation diagnosed?
Testing for the mutation involves direct (Sanger) sequencing of blood or salivary DNA and direct sequencing of tumour tissue. BAP1-inactivated melanocytic tumour tissue or other tumour tissue can be screened for loss of BAP1 nuclear staining on IHC, as it is highly sensitive and specific [27]. However, there is a risk of false negatives. The loss of two BAP1 alleles is required for the test to show a complete lack of BAP1 expression. Melanocytic naevi in germline BAP1-mutant individuals may not be BAP1 deficient on IHC staining as they retain one wild-type allele. Cells with mono-allelic inactivation will have nuclear staining but may also show cytoplasmic staining for BAP1 if the nuclear localisation sequence is affected [2]. The selection of a naevus looking like a BAP1-inactivated melanocytic tumour or a positive internal control may reduce this risk.
Note that BAP1-deficient naevi can be present in individuals without the germline mutation (due to a somatic cause with two hits confined to tumour tissue). Formal genetic testing is always required to diagnose the germline mutation.
What is the management for the germline BAP1-positive patient?
There are no formal or evidence-based cancer surveillance guidelines for patients with the germline BAP1 mutation. Currently proposed surveillance recommendations include screening for uveal melanoma, cutaneous melanoma, renal cell carcinoma, and mesothelioma [1,3,5,20,28–30].
Uveal melanoma
A yearly review with an ophthalmologist specialising in uveal melanoma is recommended, starting at 16 years of age (a younger age may be considered if a family member has been diagnosed with early uveal melanoma). Some suggest starting screening at 11 years of age, which is five years younger than the earliest reported BAP1-associated uveal melanoma [3,28]. Six-monthly ophthalmic screening is recommended from 30 years old. The examination should consist of indirect ophthalmoscopy with a well-dilated pupil, wide-field fundus photography, and ocular ultrasound.
Cutaneous melanoma
A twice-yearly full-skin examination should be undertaken by a dermatologist specialising in melanoma, involving a systematic review of total body photography from 18 years old. Sequential digital dermoscopic examination or the excision of any mildly irregular naevus or BAP1-inactivated melanocytic tumour-like lesion is recommended. Sun protection should also be emphasised.
Renal cell carcinoma and mesothelioma
Annual screening should begin between 30 and 55 years old with an abdominal and respiratory clinical examination looking for abdominal masses, distention, ascites, or signs of a pleural effusion.
- The patient may have the option of investigation of symptoms or asymptomatic imaging surveillance every two years. Imaging involves ultrasound of the chest and renal tract and consideration of magnetic resonance imaging (MRI) to investigate pleural or peritoneal disease.
- After age 55, consider adding abdomen/chest computed tomography (CT) with contrast every two years as an alternative to MRI.
- Consider following the protocol established for von Hippel–Lindau disease, which involves yearly abdominal/renal ultrasound and CT or MRI every two years [3].
- Smoking and exposure to asbestos must be avoided.
What are future treatment options for advanced BAP1-related cancers?
The therapeutic benefit of drugs targeting different parts of the BAP1 pathway is under investigation for BAP1-mutant cancers. There are currently Phase II and III clinical trials of the histone deacetylase (HDACi) inhibitor vorinostat and the enhancer of zeste homolog 2 (EZH2) inhibitors (eg, tazemetosat) for patients with advanced pleural mesothelioma and metastatic uveal melanoma. Loss of BAP1 leads to abnormal ubiquitination of histone H2A and over-expression of EZH2, which HDAC and EZH2 may counteract, respectively [5].
Acknowledgements
We would like to thank clinical geneticist Dr Annabel Goodwin, A/Prof R Max Conway, and Dr Rony Kapoor for their discussion about cancer incidence and surveillance issues in this cohort, and co-authorship on recent BAP1 surveillance proposal [30].