What is juvenile idiopathic arthritis?
Juvenile idiopathic arthritis (JIA) encompasses all chronic childhood arthropathies beginning before 16 years of age with persistent arthritis occuring for at least 6 weeks.
JIA is recognised as having seven major subtypes, some of which exhibit the clinical and pathological features of other autoimmune disorders; these subtypes include:
- Systemic JIA (also known as juvenile-onset Still disease)
- Oligoarthritis
- Rheumatic factor (RF)-positive polyarthritis
- RF-negative polyarthritis
- Psoriatic arthritis
- Enthesitis-related arthritis
- Undifferentiated arthritis [1,2].
Juvenile idiopathic arthritis
Who gets juvenile idiopathic arthritis?
JIA can occur in anyone from infants to adolescents, and has a mean onset age of 6 years of age. Most types of JIA affect boys and girls equally, although systemic-onset JIA (juvenile-onset Still disease) is predominantly seen in girls [3].
What causes juvenile idiopathic arthritis?
The exact cause of JIA is unclear, but a pronounced immune response in a perpetuating loop of both innate and adaptive immunity is believed to contribute to the pathogenesis of JIA. Aberrant activation of the innate immune system lead to the dysregulated production of proinflammatory cytokines. Tumour necrosis factor alpha (TNF-α), interleukin (IL)-1, and IL-6 play a critical inciting role. The effects of these cytokines provide explanations for the various clinical features seen in systemic JIA. These effects include:
- Bone marrow granulopoiesis (proliferation of granulocytes)
- Increase in osteoclast activity (cells that absorb bone)
- Hepatocyte stimulation
- Activation of thermoregulatory functions (body temperature) [3,4].
The increased recruitment of inflammatory cells in the joint synovial membrane was demonstrated in several studies by a significant elevation of proinflammatory cytokines in the synovial fluid of affected children. Proinflammatory cytokines in the synovium lead to increased production and accumulation of synovial fluid and thickening of the synovial lining. This chronic synovial inflammation is apparent and common to all JIA subtypes. In addition, RANKL (receptor activator of nuclear factor kappa-Β ligand) cytokine, which is associated with bone resorption (breakdown) and cartilage damage, was reported to be present in large amounts in the synovial membrane of children with JIA.
Some studies have consistently supported associations between enthesitis-related JIA and human leukocyte antigen (HLA) polymorphisms; there appear to be increased associations, especially with early-onset disease. A monogenic form of systemic JIA with an underlying mutation in a regulatory protein involved in macrophage metabolism has been described [2,3].
Environmental factors may contribute to the development of JIA. Some studies have suggested associations with exposure to antibiotics, a bacterial infection in immunosuppressed individuals, and maternal smoking during pregnancy [2,4].
What are the clinical features of juvenile idiopathic arthritis?
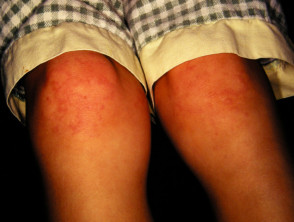
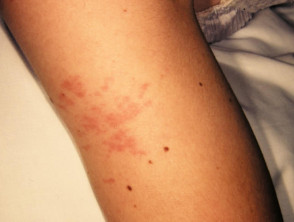
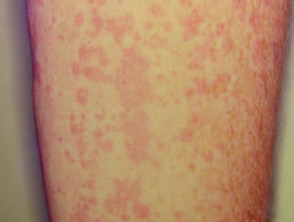
Cutaneous involvement is one of the key extra-articular factors in systemic JIA. The exanthem or rash presents as discrete, salmon-pink macules, or oedematous papules that are typically transient and coincide with fevers. These tend to occur on the trunk, axillae, and proximal extremities but are not commonly seen on the palms, soles, or the face. Persistent plaques, periorbital oedema, or erythema are less common but may be observed.
Although rashes in JIA are seldom pruritic, they can be elicited by cutaneous injury or scratching (Koebner phenomenon), in which case the lesions exhibit a linear appearance.
Children with JIA often present with limping and stiffness, without any known injury or prior infections. Common non-cutaneous manifestations include:
- Joint pain and swelling
- High spiking daily fever (quotidian pattern)
- Myalgia
- Limited movement
- Enthesitis
- A sore throat
- Lymphadenopathy
- Hepatosplenomegaly [1–3].
What are the complications of juvenile idiopathic arthritis?
Musculoskeletal complications may occur secondary to ongoing inflammation and poor disease control, including:
- A leg-length discrepancy
- Joint erosion
- Sacroiliac joint and spine ankylosis (stiffening due to bone fusion).
Sometimes JIA may result in more severe complications with devastating outcomes. These may include:
- Uveitis — this may lead to vision loss; screening and close monitoring by a paediatric ophthalmologist is warranted for severe, active uveitis
- Macrophage activation syndrome (MAS) — this may be life-threatening; it is characterised by fever, cytopenias, liver dysfunction, coagulopathy, purpura, hypofibrinogenaemia (a blood clotting factor disorder), hypertriglyceridaemia (high levels of triglycerides in the blood), and a very high level of ferritin [2,3].
How is juvenile idiopathic arthritis diagnosed?
The diagnosis of systemic JIA is made clinically after exclusion of other entities, such as malignancies and infections that can also present with high fever and arthralgia. According to the International League of Associations for Rheumatology (ILAR) criteria for systemic JIA, arthritis must have persisted for at least 6 weeks and fever for 2 weeks, accompanied by one or more of the following:
- Transient erythematous rash
- Generalised lymphadenopathy
- Hepatosplenomegaly
- Serositis (inflammation of a serous membrane) [5].
When JIA is suspected, paediatric rheumatologists should be involved promptly to initiate the appropriate investigations and to minimise any delay in treatment. Further investigations including laboratory tests and imaging are helpful to confirm the diagnosis or to classify arthritis into a specific subtype. The test results can also be useful to guide treatment and to monitor the disease course.
The most relevant laboratory tests for JIA are:
- A complete blood count — anaemia, thrombocytosis, and leucocytosis are often present in systemic JIA whereas they are not typically seen in oligoarticular JIA
- Erythrocyte sediment rate (ESR) — this is significantly elevated in systemic JIA but may be normal or mild to moderately elevated in other subtypes
- C-reactive protein (CRP) — this is elevated to varying degrees in the different subtypes of JIA
- Antinuclear antibody (ANA) — this is often negative in systemic JIA but may be positive in oligoarticular and polyarticular forms of JIA
- RF and anti-cyclic citrullinated peptide (anti-CCP); these are useful to identify RF-positive polyarthritis.
Pathology is not required for a definitive diagnosis of JIA, but the exanthem in systemic JIA characteristically exhibits a perivascular and interstitial neutrophil-dominant infiltrate; this can also be referred to as neutrophilic urticarial dermatosis, a common pattern in autoinflammatory processes [3].
Imaging can provide information about the involved joints if the clinical examination findings are equivocal. An ultrasound scan can assess synovitis in the joints, and magnetic resonance imaging may be used to rule out other conditions such as pigmented villonodular synovitis (inflammation of the joint lining) [2].
What is the differential diagnosis for juvenile idiopathic arthritis?
Several conditions may present similarly to systemic-onset JIA, including having a transient exanthem, fever, and arthritis. These include:
- Rheumatic fever
- Urticarial vasculitis
- Serum sickness-like reaction
- Juvenile dermatomyositis
- Systemic lupus erythematosus
- Kawasaki disease [3].
What is the treatment for juvenile idiopathic arthritis?
Treatment of systemic JIA is directed at symptom management, improving or managing joint function, and minimising complications. Non-steroidal anti-inflammatory drugs (NSAIDs) are typically the first-line agents for mild articular or extra-articular presentations to control pain. Other pharmacological agents include:
- Oral corticosteroids for moderate to severe disease.
- Intravenous corticosteroids — these may be required in patients with severe flare-up or complications, such as MAS.
- Disease-modifying anti-rheumatic drugs (DMARDs), such as methotrexate, ciclosporin, and leflunomide.
Drugs that block inflammatory cytokines, such as TNF-α, IL-6, and IL-1, have proven efficacy, especially in longstanding JIA. These include:
- Anakinra — an IL-1 receptor antagonist
- Canakinumab — a humanised monoclonal antibody directed against IL-1
- Tocilizumab — this humanoid monoclonal antibody blocks IL-6 [2–4].
What is the outcome for juvenile idiopathic arthritis?
The disease course of systemic JIA is variable. Reports have indicated that 40–60% of patients reach the remission of arthralgia. Systemic manifestations may remain active and may persist after years. A better prognosis is associated with the early treatment of active disease [2,3].