What is lymphomatoid granulomatosis?
Lymphomatoid granulomatosis is a rare form of B-cell lymphoma (a cancer of lymph tissue). It is characterised by lymphocytes around blood vessels, that is, they are angiocentric.
By World Health Organization definition lymphomatoid granulomatosis is defined as an angiocentric and angiodestructive lymphoproliferative disease involving extranodal sites, composed of B cells positive for Epstein-Barr virus (EBV) and admixed with reactive T cells. It also often occurs in association with an underlying immunodeficiency state. It is a form of non-Hodgkin lymphoma. See Epstein–Barr virus-associated lymphoproliferative disorders.
Lymphomatoid granulomatosis is twice as common in males as in females and occurs between 30 and 50 years of age. The lungs are generally involved with the skin and central nervous system also commonly affected. Other organs can be involved including kidney, liver, spleen, lymph nodes, eyes and the gastrointestinal tract.
What are the symptoms?
The most common presenting symptoms are cough, fever and skin lesions. Affected patients may feel generally unwell, with weight loss, neurological abnormalities, breathlessness and chest pain.
What are the skin examination findings?
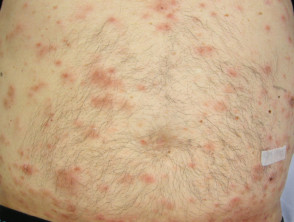
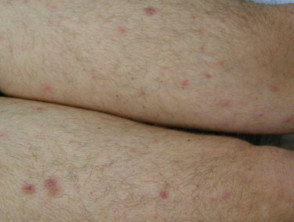
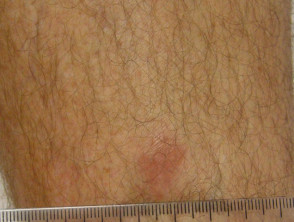
The skin lesions can be in the form of:
- Small red bumps (papules)
- Lumps under the skin (nodules)
- Ulcers
- Thickened patches
- Inflamed hair follicles (folliculitis).
There are usually no symptoms from the skin lesions, but they can be tender or itchy. Neurological examination may reveal cranial and peripheral nerve defects resulting in unsteadiness, blurred vision, weakness or numbness affecting facial muscles or hands and feet.
Lymphomatoid granulomatosis
What tests may be performed?
Tissue biopsy is needed to confirm a diagnosis of lymphomatoid granulomatosis. The results of other laboratory tests tend to be non-specific.
What are the characteristic histopathological findings?
Biopsy findings in lymphomatoid granulomatosis include a classic triad of features.
- Mixed lymphoid infiltrate
- Lymphocytes infiltrating through the walls of arteries and veins
- Focal areas of necrosis (cell death).
Special stains (immunohistochemistry) show large numbers of reactive T-cells with varying numbers of malignant B-cells. The histological grading is from 1-3 (low to high). Grade 1 only has small numbers of EBV-positive B cells. Grade 2 has increasing numbers of large EBV-positive B cells. Grade 3 more closely resembles conventional forms of diffuse large B-cell lymphoma.
What are the associated immunodeficiency states?
Lymphomatoid granulomatosis occurs in the setting of a poorly functioning immune system, for example:
- Organ transplantation
- Wiskott-Aldrich syndrome
- Human immunodeficiency virus (HIV) infection
- X-linked lymphoproliferative syndrome
- Common variable immunodeficiency syndrome
- Other reduced immune surveillance disorders.
What are the treatment options?
The ideal treatment for lymphomatoid granulomatosis is not known. Treatments that have been reported to be successful in at least some patients include:
- Systemic steroids plus cyclophosphamide
- Interferon alpha 2b
- Combination chemotherapy with CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone)
- Anti-CD20 monoclonal antibodies (rituximab)
- Anti-CD20 monoclonal antibodies + CHOP
- Autologous haematopoietic stem cell transplantation after high-dose chemotherapy.
What is the prognosis with lymphomatoid granulomatosis?
Spontaneous recovery occurs in about 20% of patients with low-grade disease (grade 1). Others have progressive disease. Reported 5-year mortality rates have been between 38% and 88%. The median survival is between 14 and 72 months. Prognosis is worse with grade 2 and 3 disease and if there is neurological involvement.