What is Sneddon syndrome?
Sneddon syndrome is a rare, slowly progressive, neurocutaneous vasculopathy. It is characterised by the combination of livedo racemosa and recurrent cerebrovascular events, such as transient ischaemic attacks and strokes.
Sneddon syndrome is also called Ehrmann-Sneddon syndrome, livedo racemose and cerebrovascular accident syndrome, and livedo reticularis and cerebrovascular accident syndrome.
Who gets Sneddon syndrome?
The incidence of Sneddon syndrome is approximately four per million per year in the general population. Sneddon syndrome is typically first diagnosed in women aged 20–42 years. There does not appear to be any racial variation, although reported numbers are small.
Sneddon syndrome is often associated with autoimmune diseases, such as antiphospholipid syndrome and systemic lupus erythematosus.
What causes Sneddon syndrome?
As antiphospholipid antibody markers are present in 50–80% of patients with Sneddon syndrome, it is included in the clinical spectrum of primary antiphospholipid syndrome.
Vascular thrombosis and recanalisation of small and medium-sized blood vessels in the skin and brain are due to coagulopathy or thrombophilia, which is associated with decreased levels of activated protein C and protein S and elevated levels of factor VII.
Sneddon syndrome can be inherited as an autosomal dominant disease with variable penetrance in rare familial cases. Loss-of-function mutations have been identified in the cat eye syndrome region 1 candidate 1 (CECR1) on chromosome 22, encoding adenosine deaminase 2 (ADA2).
What are the clinical features of Sneddon syndrome?
Sneddon syndrome has dermatological and neurological manifestations.
Dermatological manifestations
Livedo racemosa and acrocyanosis
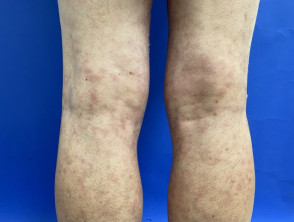
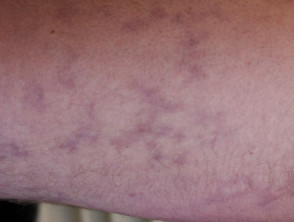
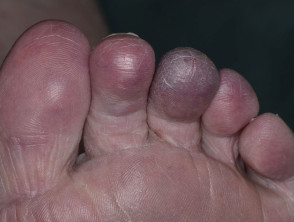
Sneddon syndrome is characterised by livedo racemosa, which precedes the onset of recurrent strokes by over 10 years [1,2]. The significance of the livedo racemosa, which often begins in childhood, is only recognised in the 20s and 30s after the cerebrovascular events begin.
- Livedo racemosa is due to persistent obstruction of peripheral blood flow due to the occlusion of small or medium-sized arteries.
- It presents as a branching, net-like pattern of dusky erythematous or violaceous broken circles.
- Unlike livedo reticularis, skin discolouration remains unchanged with warming, although it is more prominent during cold exposure and pregnancy.
- Livedo racemosa initially affects the buttocks and lower back.
- It gradually progresses to involve the dorsal aspects of the thighs and arms.
- It rarely affects the face, hands, or feet.
- Lesions are painless and are not associated with oedema, ulceration, or pruritus.
Other dermatological manifestations associated with Sneddon syndrome include:
Neurological manifestations
Neurological manifestations associated with Sneddon syndrome generally present in three stages.
- Prodromal symptoms may comprise vertigo, dizziness, and headaches.
- In the second stage, patients experience recurrent episodes of strokes or transient ischaemic attacks due to ischaemia in the zones perfused by the middle or posterior cerebral arteries. Typical symptoms include aphasia, visual field defects, hemiparesis, and sensory disturbances.
- The third stage is characterised by significant cognitive decline and early-onset dementia, due to the cumulative effect of multiple cerebral infarcts. Cognitive dysfunction, affecting memory, attention, and visuospatial domains, and psychiatric disturbances, such as depression, are reported in approximately 77% of patients with Sneddon syndrome.
Cerebrovascular complications of Sneddon syndrome include:
- Ischaemic stroke
- Haemorrhagic stroke (rare)
- Seizures
- Movement disorder (rare), such as chorea or cerebellar tremor.
What are the complications of Sneddon syndrome?
Cardiovascular complications of Sneddon syndrome include:
- Mitral or aortic valvulopathy
- Libman-Sacks endocarditis
- Systolic labile hypertension.
Ophthalmological complications of Sneddon syndrome include:
- Optic disc microaneurysm
- Macular oedema
- Retinal artery branch obstruction.
Renal complications are rare:
- Impaired creatinine clearance
- Renal artery thrombosis.
How is Sneddon syndrome diagnosed?
Criteria for diagnosis of Sneddon syndrome include the presentation of livedo racemosa with characteristic histopathological findings on skin biopsy and focal neurological deficits. History of transient ischaemic attacks or stroke or evidence of these on imaging can support the diagnosis.
Patients with a suspected diagnosis of Sneddon syndrome should have blood tests screening for autoimmune disease and coagulopathies, cerebral MRI, cardiovascular assessment, and skin biopsy.
Lesions identified by MRI tend to be small, multifocal, and often located in the periventricular deep white matter or pons. In up to 75% of patients with Sneddon syndrome, cerebral angiography is abnormal.
On deep skin biopsy, histopathology demonstrates a non-inflammatory thrombotic vasculopathy involving subcutaneous small and medium-sized dermal arteries. Sensitivity increases with the number of deep skin biopsies: 27% sensitivity with one, 53% with two, and 80% with three biopsies.
Extensive investigation is necessary for patients presenting with livedo racemosa or stroke to rule out other diseases in the differential.
Classification
Sneddon syndrome is classified as antiphospholipid negative or positive, based on the presence of any of 3 antiphospholipid antibodies: anticardiolipin, lupus anticoagulant, and anti-beta 2-glycoprotein I [2].
It can also be classified as primary (idiopathic) or secondary to an identified thrombophilia or autoimmune disease, such as lupus erythematosus [1]. Sneddon syndrome is also occasionally classified as a vasculitis.
What is the differential diagnosis for Sneddon syndrome?
The differential diagnosis for Sneddon syndrome may include:
- Congenital livedo reticularis
- Another haematological condition
- Antiphospholipid syndrome
- Cryoglobulinaemia
- Polycythaemia vera
- Autoimmune disease
- Cardiac disease resulting in multiple emboli
- Neoplasia
- Lymphoma
- Leukaemia
- Infection
- Effects of a drug
- Amantadine
- Minocycline
- Quinidine
- Divry van Bogaert Syndrome.
What is the treatment for Sneddon syndrome?
Treatment is based on anecdotal reports and includes:
- Long term anticoagulation — in patients positive for antiphospholipid antibodies, warfarin is recommended. Patients without antiphospholipid antibodies may benefit from antiplatelet therapy
- Prostaglandin E1
- Angiotensin-converting enzyme (ACE) inhibitors
- Nifedipine – for Raynaud phenomenon
- Immunosuppressive drugs, such as cyclophosphamide.
Lifestyle modifications recommended for Sneddon syndrome include:
- Cessation of smoking
- Cessation of oestrogen-containing oral contraceptives
- Reduction of modifiable cardiac risk factors.
What is the outcome for Sneddon syndrome?
Sneddon syndrome is a chronic, progressive disease often resulting in cognitive decline and early-onset dementia due to the cumulative effect of multiple cerebral infarcts.