What is Waardenburg syndrome?
Waardenburg syndrome is a rare genetic disorder characterised by sensorineural hearing loss and pigmentary abnormalities of the hair, skin, and eyes.
Waardenburg syndrome is named after Petrus Johannes Waardenburg, a Dutch ophthalmologist, who noticed that heterochromia iridis often accompanied deafness.
Waardenburg syndrome
Who gets Waardenburg syndrome?
Waardenburg syndrome may be inherited or new mutations can arise spontaneously. It affects males and females equally, and can affect all races.
It has a population frequency of 1 in 40,000, and is responsible for 2–5% of all cases of congenital deafness.
What causes Waardenburg syndrome?
Waardenburg syndrome is a neurocristopathy due to gene mutations which result in abnormal neural crest differentiation during embryonic development.
Mutations in a number of different genes can cause Waardenburg syndrome, with some differences in symptoms and signs. Expression and penetrance are also variable.
- WS1 and WS3 are caused by mutations in the PAX3 gene at chromosome 2q
- WS2A – MITF (microphthalmia associated transcription factor) gene, chromosome 3p
- WS2B – chromosome 1p
- WS2C – chromosome 8p
- WS2D – SNAI2 gene, chromosome 8q
- WS2E – SOX10 gene, chromosome 22q
- WS4A – EDNRB gene at 13q
- WS4B – EDN3 gene at 20q
- WS4C – SOX10 gene (as for WS2E)
The mutations that cause Waardenburg syndrome include insertions, deletions, frameshifts, splice alterations, missense, or nonsense mutations.
Most types of Waardenburg syndrome are autosomal dominant, meaning that only one affected gene needs to be passed on to a child for them to have the syndrome. Transmission of defects in EDN3 or EDNRB is more complex; they are usually autosomal recessive, although cases of autosomal dominant transmission with incomplete penetrance have been described.
Other mutations in some of the above genes can cause related clinical syndromes, such as Tietz syndrome (MITF gene), piebaldism (SNAI2 gene), PCWH (SOX10 gene), or ABCD syndrome (EDNRB gene).
What are the clinical features of Waardenburg syndrome?
Features are present from birth. As it is a rare condition and clinical signs can be subtle, diagnosis may not be made until later in life. In addition to the characteristic pigment changes and deafness, Waardenburg syndrome can be associated with musculoskeletal defects and Hirschsprung syndrome.
Four major clinical types of Waardenburg syndrome have been identified.
Type 1
Type 1 is the most common subtype of Waardenburg syndrome. Features include:
- Sensorineural deafness
- Typical facial structure, with dystopia canthorum (lateral displacement of the medial corners of the eyes), broad nasal root, and synophrys (meeting of the eyebrows in the midline)
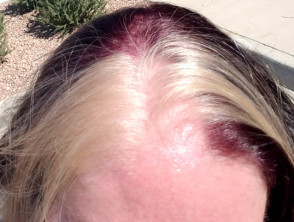
- White skin patches (leukoderma) and white forelock of hair (poliosis)
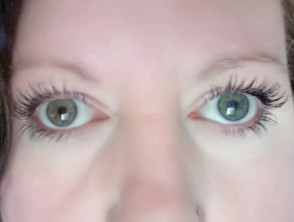
- Pigmentary abnormalities of the eyes are less common than the skin and hair changes; heterochromia iridis (differently coloured eyes), which may be complete, partial, or segmental; isohypochromia iridis (pale blue eyes); or abnormal pigmentation of the fundus
Type 2
Type 2 has similar clinical features to type 1 Waardenburg syndrome, but the inner canthi are normal.
Type 3
Type 3 (Klein-Waardenburg syndrome) also has similar features to type 1 Waardenburg syndrome, but with musculoskeletal abnormalities, such as muscle hypoplasia, flexion contractures, or syndactyly (fused digits).
Type 4
Type 4 (Shah-Waardenburg syndrome) has similar features to type 2 Waardenburg syndrome but with Hirschsprung syndrome (a condition resulting from missing nerve cells in the muscles of part or all of the large intestine).
How is Waardenburg syndrome diagnosed?
Waardenburg syndrome is diagnosed on clinical features. In 1992 the Waardenburg Consortium developed major and minor criteria for diagnosis.
Major criteria
- Sensorineural deafness
- Iris pigmentary abnormality, such as heterochromia iridis – complete, partial, or segmental; isohypochromia iridis; or fundus pigmentary abnormalities
- Abnormalities of hair pigmentation, such as white forelock, eyebrows, or eyelashes (poliosis)
- Dystopia canthorum – lateral displacement of inner canthi
- First degree relative with Waardenburg syndrome
Minor criteria
- Unpigmented patches of skin (leukoderma)
- Synophrys
- Broad nasal root
- Hypoplasia of alae nasi
- Premature greying of scalp hair
A clinical diagnosis of type 1 Waardenburg syndrome needs 2 major, or 1 major and 2 minor criteria.
The W index
The W index can be calculated to determine whether dystopia canthorum is present.
- a=inner canthal distance
- b=interpupillary distance
- c=outer canthal distance
- X=(2a-(0.2119c+3.909))/c
- Y=(2a-(0.2479b+3.909))/b
- W=X+Y+a/b
- W index > 1.95 is indicative of dystopia canthorum.
Genetic sequencing of the PAX3 gene for mutations causing Waardenburg syndrome may be performed as part of genetic counselling of family members. Prenatal testing for PAX3 mutations is possible by chorionic villus sampling or amniocentesis, but is rarely done due to the clinical variation found within Waardenburg syndrome and the mutation will not indicate which clinical features will be present or their severity.
What is the treatment for Waardenburg syndrome?
As Waardenburg syndrome is a genetic disease there is no curative treatment. Genetic counselling may be helpful for affected patients who want to start a family.
- Audiology examinations should be performed for children with suspected Waardenburg syndrome to evaluate them for deafness. Hearing aids or cochlear implants may be needed.
- Patients with Hirschsprung syndrome may require surgery to remove the affected segment of bowel.
- Unpigmented patches of skin are more susceptible to sun damage, so sun protection is important.
What is the outcome for Waardenburg syndrome?
The clinical features of Waardenburg syndrome are stable and the features will remain throughout life. Life expectancy is normal.