Introduction - Langerhans cells
Introduction
Demographics
Classification
Diagnosis
Treatment
Prognosis
Langerhans cells are immune cells that are normally found within the epidermis where they act as antigen-presenting cells in an early warning system fighting foreign material such as bacteria. They may migrate to the local lymph glands but usually return to the skin.
Langerhans cell histiocytosis (LCH) refers to a reactive increase in the number of Langerhans cells in the skin and other organs (see histiocytoses). Langerhans cell histiocytosis may also be called ‘class I histiocytosis’ or ‘histiocytosis X’.
Langerhans cell histiocytosis is a rare condition, slightly more common in boys than girls, affecting around one in 5 million children and fewer adults. Langerhans cell histiocytosis may develop at any age but most commonly occurs in childhood (1–3 years of age).
There is a range of ways this disease may present and some of the well-recognised patterns of this disease have been given separate names:
All of these subtypes overlap and the skin disease is similar for all of them. It generally presents with many small pinkish or reddish-brown papules on the skin that may become crusted and infected.
Recent information suggests that Langerhans cell histiocytosis may be a reaction to an underlying viral infection. In at least some cases it has been associated with Epstein-Barr virus.
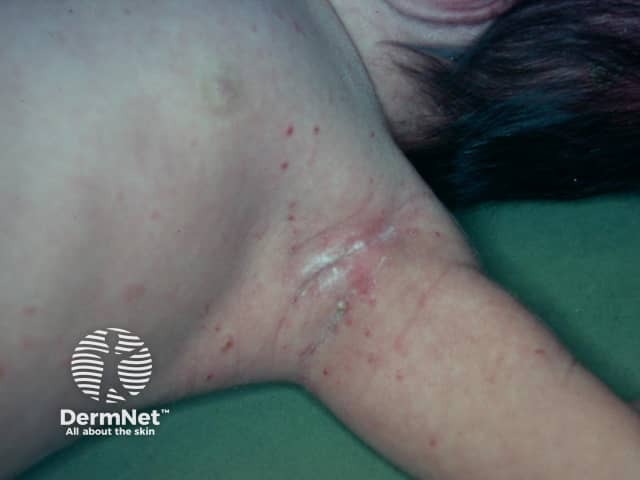
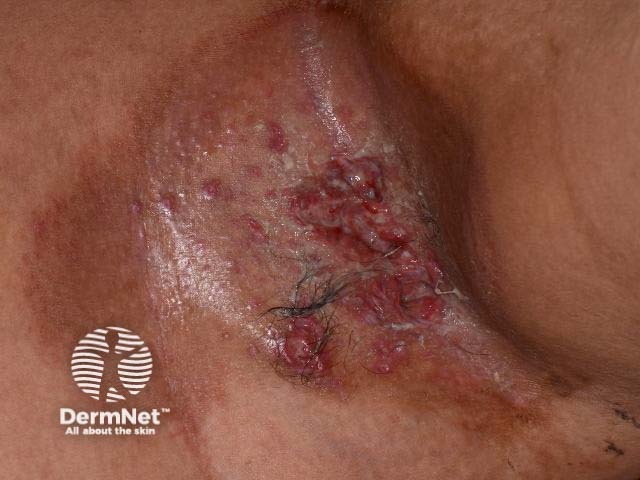
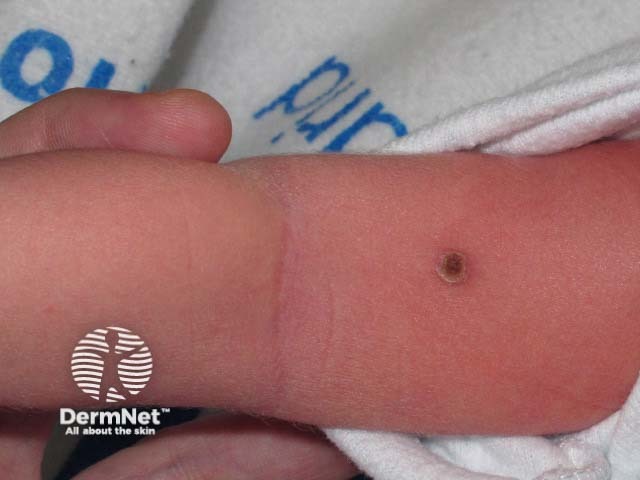
The appearance of the rash or X-rays and scans of internal organs may give the doctor a clue to the diagnosis. A skin biopsy of the affected area is usually needed to confirm the diagnosis. The biopsy findings do not give any indication of the likely course of the disease.
The diagnosis may be obvious when the biopsy tissue is looked at under a microscope, but often special stains are needed to clearly identify the type of histiocytosis.
When histiocytosis is diagnosed in one organ (eg, the skin or bone) other organs such as the blood, lungs, liver, and kidneys are checked to determine whether they are also affected by histiocytosis.
Treatment depends on the severity of the disease and the number of organs that are involved. Evidence of damage to the organs is more important than involvement of the organ as such.
Disease limited to the skin only may not require treatment if mild. If treatment is necessary due to discomfort or cosmetic concerns the following treatments may be used:
The disease affecting limited areas of the bone may be treated if causing pain or if there is a risk of deformity or fracture. The limited bone disease may be treated with:
When more than one organ (eg, skin and bone) is damaged chemotherapy is usually considered. Children are usually tried on oral corticosteroids first, progressing to chemotherapy if this is ineffective. It is not clear at this time which is the most effective and safe form of chemotherapy. Decisions about chemotherapy are based on the likely long-term prognosis for an individual person.
Not all people with multiple organ involvement will respond to treatment.
Many people may have mild disease confined to one organ with few or no symptoms. These people have a good prognosis.
Others may have more severe disease involving many organs within the body. This can be fatal. The predictors of a poor outcome are:
Children less than two years of age with LCH affecting many organs have a mortality rate of 40–50%. It is uncommon for severe LCH affecting many organs to occur in adults. People with a single affected organ or limited bone and skin disease tend to do better but the condition may still last for many years and can become worse over time.