What is Wiskott-Aldrich syndrome?
Wiskott-Aldrich syndrome (WAS) is a rare, inherited immune deficiency disorder that results in infections and is also associated with microthrombocytopenia (low platelet count and abnormally reduced platelet size), eczema, an increased risk of autoimmune diseases and some types of cancer.
The syndrome is due to mutations or deletions in a gene found on the X chromosome that codes for Wiskott-Aldrich Syndrome Protein (WASP). The WAS protein is pivotal in signalling and the cytoskeletal (structural) organization of haemopoietic (blood) cells. Different types of mutation within the WASP gene may vary between individuals resulting in the presence of the complete clinical spectrum in some cases, or only some of the features in others.
Approximately 160 different mutations or deletions in the WASP gene have been described. Because the gene coding for WASP is X-linked, the majority of cases with Wiskott-Aldrich syndrome are males; rarely girls can be affected. Wiskott-Aldrich syndrome is estimated to occur in approximately 1 to 10 of every million boys. Females with the abnormal gene are usually unaffected carriers that pass the mutation on to the next generation.
How is Wiskott-Aldrich syndrome classified?
Wiskott-Aldrich syndrome can be thought of as a spectrum presenting with features that fall between the severe features of classic Wiskott-Aldrich syndrome and the less severe form called X-linked thrombocytopenia (XLT). Patients may shift in severity, depending on the progression of the disease over time, or the emergence of complications such as the development of lymphoma.
What are the symptoms of Wiskott-Aldrich syndrome?
Thrombocytopenia may be present from birth and cause prolonged bleeding from the umbilical cord. Thrombocytopenia also causes petechiae (pinpoint bleeding into the skin) and bruises (ecchymoses) which may occur without injury. Thrombocytopenia may also result in oral, and nose bleeds intestinal and intracranial bleeding. Bleeding may be life-threatening.
Eczema affects 80% of patients with Wisckott-Aldrich syndrome. Typically eczema appears during infancy or early childhood. The features of the eczema are not distinguishable from atopic eczema. Wiskott-Aldrich syndrome patients often have elevated IgE levels and develop allergies.
Immunodeficiency may affect both T and B lymphocyte function. Immune deficiency increases the risk and frequency of a wide range of infections including discharging ears, bacterial or viral pneumonia, bacterial skin infections and herpes simplex (cold sore virus) infections. Opportunistic lung infection with Pneumocystis jiroveci can occur.
Wiskott-Aldrich syndrome can be associated with a wide range of autoimmune diseases, most commonly autoimmune haemolytic anaemia, cutaneous vasculitis, arthritis and kidney disease. Affected patients may have multiple autoimmune diseases concurrently.
The commonest malignancies associated with Wisckott-Aldrich syndrome are leukaemia and B-cell lymphoma.
What are the skin findings in Wiskott-Aldrich syndrome?
There is a range of skin findings in Wiskott-Aldrich syndrome:
- Wiskott-Aldrich syndrome causes acute and chronic eczema indistinguishable from atopic eczema. Its severity and persistence are variable; in its most severe form, it is resistant to many of the common eczema treatments available.
- Petechiae and bruises may occur secondary to scratching of eczematous skin or spontaneously on unscratched skin.
- Opportunistic skin infections such as molluscum contagiosum, herpes simplex and bacterial sepsis may also be persistent or recurrent problems for Wiskott-Aldrich syndrome patients.
Genetics of Wiskott-Aldrich syndrome*
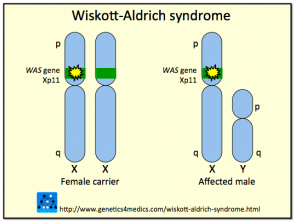
*Image courtesy Genetics 4 Medics
What tests may be used in the diagnosis of Wiskott-Aldrich syndrome?
On full blood count testing, the platelet count is almost always low, and the platelets are characteristically smaller than normal. In some cases, the platelet count is within the normal range, but platelet size is always affected. The neutrophil and lymphocyte count may also be low.
If an infant or child with eczema has signs of thrombocytopenia and suspected immunodeficiency, they should have a full blood count performed. If there is thrombocytopenia or small platelets are seen on the blood film, a referral to a paediatrician is recommended.
If there is strong clinical suspicion of Wiskott-Aldrich syndrome and there are full blood count abnormalities, the case should always be discussed with a paediatric immunologist before undertaking further investigations.
Immunoglobulin (antibody) levels in the bloodstream may be low in Wiskott-Aldrich syndrome. Classic Wiskott-Aldrich syndrome is associated with:
- low immunoglobulin M (IgM) and immunoglobulin G (IgG) levels
- normal-to-high immunoglobulin A (IgA) and immunoglobulin E (IgE) levels.
However, young infants, in particular, may not show classic immunoglobulin abnormalities because Wiskott-Aldrich syndrome is associated with a gradual decline in immunological function.
Immunoglobulin responses to vaccination may be absent, especially responses to pneumococcal vaccines.
The confirmatory diagnostic test for Wiskott-Aldrich syndrome is genetic testing of blood lymphocytes to identify whether there is a specific mutation of or deletion within the WASP gene.
What are the treatment options for Wiskott-Aldrich syndrome?
Haemopoietic stem cell transplant, which is usually a bone marrow transplant, is curative for Wiskott-Aldrich syndrome patients. For those with a genetically compatible sibling donor (about 20% of patients), there is an 80% survival rate, and the survival rate is only slightly less for an unrelated matched donor transplant. The survival rate is less and complications more frequent for mismatched related donor transplant. The outcome is improved if haemopoietic stem cell transplant is undertaken early in childhood.
Before transplantation or if transplantation is not an option, affected children may need specific treatment for immunodeficiency and bleeding problems including:
- Trimethoprim-sulfamethoxazole prophylaxis daily to prevent Pneumocystis jiroveci infection
- Aciclovir prophylaxis to prevent herpes simplex infections
- Regular intravenous immunoglobulin infusions where there are abnormalities in B cell function
- Irradiated platelets and red blood cell transfusions to treat serious bleeding episodes.
Live vaccines such as BCG and MMR are contraindicated. Non-steroidal anti-inflammatory medications (ibuprofen, diclofenac, aspirin and others) should be avoided.
Gene therapy for Wiskott-Aldrich syndrome remains experimental.
What is the outlook for children with Wiskott-Aldrich syndrome?
The average life expectancy for boys with Wiskott-Aldrich syndrome is about 15 – 20 years without haemopoietic stem cell transplant. Engrafted children are expected to survive much longer.
What genetic information do families need?
Once the genetic abnormality is identified in the affected boy, his mother can be tested to see whether she carries the gene. If she does, then she has a 50% risk of having other affected sons. There is also a 50% risk that she may transmit the gene to her daughters who become carriers and may have their affected sons. Antenatal diagnosis is available for affected families.