What is lymphocytic thrombophilic arteritis?
Lymphocytic thrombophilic arteritis is a primary lymphocytic vasculitis. It presents with livedo racemosa or macular hyperpigmentation. It has distinct histological findings affecting small to medium-sized arteries in the deep dermis and superficial subcutis [1].
It is also known as macular lymphocytic arteritis.
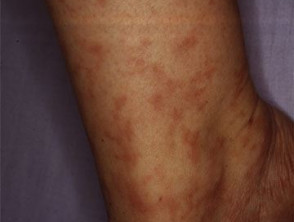
Livedo racemosa
Who gets lymphocytic thrombophilic arteritis?
Lymphocytic thrombophilic arteritis shows a female predominance, with a median age of 39 years (range 6-73) [2].
What causes lymphocytic thrombophilic arteritis?
The exact cause of lymphocytic thrombophilic arteritis is unknown. It has been proposed that the underlying lymphocytic endovasculitis drives a localised thrombophilia, represented by the intraluminal fibrin ring seen on histology, without any evidence of destruction of the vessel wall [1].
- Some cases have been associated with autoimmune antibodies or heterozygosity for prothrombotic mutations, suggesting that autoimmunological and thrombophilic factors may contribute.
- Antiphospholipid antibodies have been detected in some cases, but the low titres and lack of systemic features argue against antiphospholipid syndrome contributing significantly to its pathogenesis [1,3].
- Low to moderate levels of antinuclear antibodies (ANA) have been reported, but there was no evidence of systemic connective tissue disease in these cases [1,4].
What are the clinical features of lymphocytic thrombophilic arteritis?
The clinical features of lymphocytic thrombophilic arteritis include:
- Persistent livedo racemosa or macular hyperpigmentation typically affecting the lower limbs; the upper limbs can be affected to a lesser extent. This is non-blanching and may form an annular pattern.
- Palpable subcutaneous indurations
- Some reports of ulceration [5,6]
- Lack of itch or pain
- Absence of purpura, scarring, or atrophie blanche.
How is lymphocytic thrombophilic arteritis diagnosed?
The diagnosis of lymphocytic thrombophilic arteritis is based on a combination of typical clinical features supported by distinct histological findings [1].
- A dense inflammatory infiltrate is found in the muscular vessel wall, affecting the small and medium-sized arteries of the deep dermis, junction of the dermis and subcutis, or superficial subcutis.
- The inflammatory infiltrate consists predominantly of mononuclear cells, mainly lymphocytes and some histiocytes.
- Neutrophils and eosinophils are scant or absent.
- A concentric hyalinised fibrin ring involves the entire periphery of the lumina of the affected vessels.
Signs or symptoms of systemic vasculitis are also absent (see Cutaneous vasculitis) [2].
What is the differential diagnosis for lymphocytic thrombophilic arteritis?
Lymphocytic thrombophilic arteritis can be confused with several other vascular occlusive conditions showing livedo reticularis.
Cutaneous polyarteritis nodosa
- Cutaneous polyarteritis nodosa is characterised by painful subcutaneous nodules and ulceration, features that are uncommon in lymphocytic thrombophilic arteritis [7].
- Livedo in cutaneous polyarteritis nodosa is localised around the ulcers with an irregular starburst pattern [7].
- Early-stage cutaneous polyarteritis nodosa shows neutrophilic infiltration involving the muscular layer of medium-sized and small arteries, with more mononuclear cell involvement in the later stages [7].
Livedoid vasculopathy
- Although livedo racemosa can be present, livedoid vasculopathy typically presents in the early stages with episodic purpura and painful ulceration affecting the lower limbs, followed by atrophie blanche.
- There is luminal fibrin deposition in livedoid vasculopathy, but the inflammatory infiltrate is often scant [1,8].
Antiphospholipid syndrome
- Livedo racemosa can also be present in antiphospholipid syndrome.
- There is thrombosis without significant inflammation in the vessel wall.
- Antiphospholipid syndrome is characterised by recurrent thrombosis, miscarriages, and thrombocytopenia [9].
Sneddon syndrome
- The pattern of livedo racemosa in Sneddon syndrome is broader compared to lymphocytic thrombophilic arteritis [3].
- The livedo is more proximal in Sneddon syndrome, involving the trunk and back; the extremities are rarely involved.
- The diagnosis of Sneddon syndrome requires cerebrovascular involvement [10].
What is the treatment for lymphocytic thrombophilic arteritis?
Lymphocytic thrombophilic arteritis tends to show an indolent course with persistent asymptomatic livedo racemosa or macular hyperpigmentation [1]. As most lesions are asymptomatic, treatment is not always required [2].
Treatment options that have shown a beneficial response include:
- Aspirin
- Pentoxifylline
- Dapsone.
Oral and topical steroids have not been shown to be helpful [2].
Given the rarity of lymphocytic thrombophilic arteritis and inconsistent treatment responses, there is no current consensus on treatment recommendations.
What is the outcome for lymphocytic thrombophilic arteritis?
Lymphocytic thrombophilic arteritis tends to follow a chronic indolent course with no progression to systemic vasculitis [1,2]. Ongoing follow-up is advisable as there have been rare reports of later systemic vasculitis [11]. The long-term prognosis remains unclear.