What are multiple self-healing squamous epitheliomas?
Multiple self-healing squamous epitheliomas (MSSE) is a rare inherited skin condition characterised by the sudden appearance of multiple skin lesions resembling well differentiated squamous cell carcinomas (SCC) or keratoacanthomas (KA) clinically and histologically. However, these lesions tend to spontaneously regress, leaving pitted scars.
Other names for the disorder are:
- Familial multiple keratoacanthomas
- Ferguson-Smith multiple keratoacanthomas
- Ferguson-Smith disease.
Who gets multiple self-healing squamous epitheliomas?
MSSE was first described in 1934 by the Scottish Dermatologist J Ferguson-Smith, who reported a young man who had developed multiple self-healing squamous tumours. It was later reported in the patient’s daughter. In 1971, Malcolm A Ferguson-Smith (the geneticist son of the dermatologist) presented information on 62 cases of MSSE from the West of Scotland, including pedigrees compatible with an autosomal dominant pattern of inheritance.
In MSSE, the skin lesions tend to appear for the first time during the second or third decade of life (range 8-70 years, median 28).
Cases of MSSE have been identified in a wider geographical area, in Japan, America, and Denmark, suggesting that the disease is more common than originally thought. In some families, the phenotype is milder, and the onset of disease later than in other families.
What causes multiple self-healing squamous epitheliomas?
MSSE was initially thought to be caused by a founder mutation. Although the disease is inherited in an autosomal dominant fashion (from an affected parent to half of his/her children), it is not fully penetrant, as there are reports of obligate carriers showing no clinical symptoms. Eleven different heterozygous mutations have been identified in the Transforming Growth Factor Beta Receptor 1 (TGFBR1) gene located on Chromosome 9q22.
It is thought that wildtype TGFBR1 acts as a tumour suppressor, until somatic deletion of the wildtype gene by a classic second "hit" results in carcinogenesis.
What are the clinical features of MSSE?
The SCC/KA-like lesions arise in crops or singly, most commonly in sun-exposed areas and at sites of trauma. The nose, ears, and perioral face are the most common involved sites. The trunk is rarely involved and lesions never develop on the palms and soles. lesions on the limbs tend to be larger than those on the face.
Each lesion begins as a dull red macule that grows rapidly over 2-4 weeks to become a papule or nodule. It may reach 2–3 cm in diameter, and remain stable for up to 2 months. A central horny plug forms and falls out to leave a crateriform tumour, before involuting to a pitted scar.
Patients may have up to one hundred lesions over time, but some develop only one or two lesions in their lifetime.
Family members may develop these lesions despite having relatively little sun exposure in the past, unlike most people with SCC and KA.
The SCC/KA-like lesions in mother and daughter with familial multiple keratoacanthomas
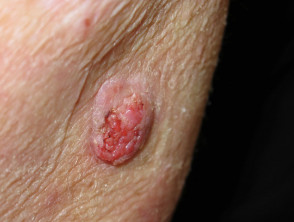
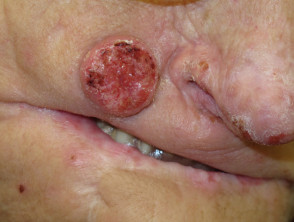
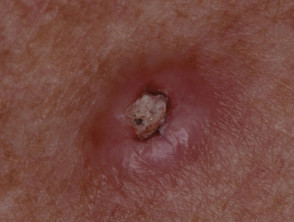
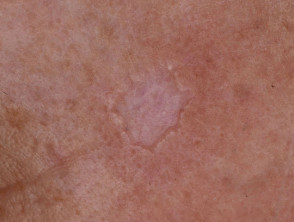
How is MSSE diagnosed?
Ferguson-Smith disease should be considered in patients presenting with multiple SCC/KA-like lesions at a young age with a positive family history. In areas of high ultraviolet (UV) exposure and skin cancer prevalence, the diagnosis of MSSE may be delayed or easily missed.
The characteristic histology of the familial SCC/KA-like lesion differs from usual keratoacanthoma, as there is no marked ‘shouldering’, and leucocyte abscesses are absent. They appear to be aggressive SCC histologically yet they usually heal by themselves. [see Squamous cell carcinoma pathology]
Patients with early onset of histologically aggressive squamous cell carcinomas and a family history of other affected members should be investigated for possible MSSE.
DNA can be extracted from blood or oral scrape to perform linkage studies and haplotype analysis, looking for mutations in the TGFBR1 gene on chromosome 9q22. A heterozygous loss-of-function mutation in this gene leads to susceptibility to familial multiple self-healing squamous epitheliomas.
What is the differential diagnosis of familial multiple self-healing squamous epitheliomas?
- Non-familial cutaneous SCC and keratocanthomas
- Keratoacanthomas and SCC in Muir-Torre syndrome
- Generalised eruptive KAs of Grzybowski syndrome
- Xeroderma pigmentosum
What is the treatment for MSSE?
Although individual SCC/KA-like lesions will generally resolve spontaneously, treatment can hasten this and also improve cosmetic outcome. Mostly, the lesions are removed surgically by excision or curettage and cautery. Other reported treatments are:
- Cryotherapy
- Intralesional bleomycin
- Intralesional 5-fluorouracil.
Radiotherapy is best avoided in MSSE, as this can lead to further development of new lesions within the treated field.
Oral retinoids such as acitretin may be used to reduce the number of tumours.
Patients with MSSE should be advised to protect their skin from exposure to the sun.
What is the outlook for MSSE?
The SCC/KA-like lesions of MSSE heal with scarring so this condition can be very disfiguring for those with severe disease. Although individual lesions can be locally invasive, distant metastases have not been reported.