Introduction - epidermolysis bullosa
Introduction
Demographics
Causes
Clinical features
Diagnosis
Differential diagnoses
Treatment
Prognosis
Epidermolysis bullosa (EB) is the name given to a group of inherited blistering diseases that are present from birth.
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disease in which tense subepithelial blisters appear at sites of trauma. Unlike EB, EBA is not inherited and usually presents in adult life.
EBA blisters tend to be localised to areas that are easily injured such as the hands, feet, knees, elbows, and buttocks. Sometimes there is mucosal involvement with blisters forming in the mouth, nose and eyes.






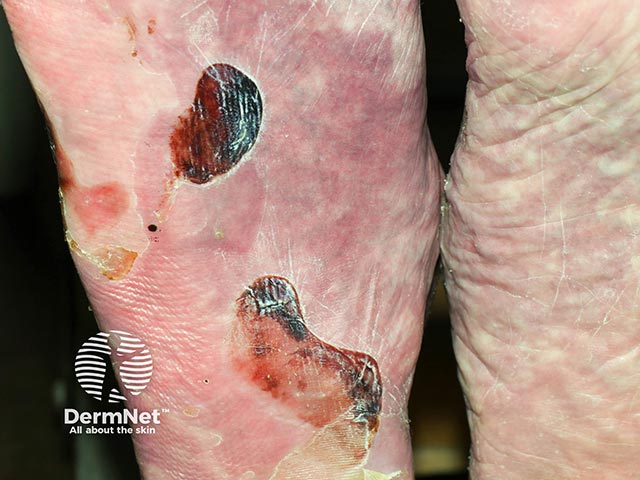
View more images of epidermolysis bullosa acquisita
EBA usually occurs in the fourth and fifth decade of life. Males and females of all races can be affected. It is estimated to have an incidence of 0.08 to 0.5 cases per million individuals; 5% of cases are reported in children, with cases reported as young as 2 weeks. Most cases present between the ages of 50⎻70 years. A neonatal case has been reported due to transfer of antibodies across the placenta.
Some patients with EBA have been reported to have other autoimmune diseases, most often Crohn disease, rheumatoid arthritis, thyroiditis, and systemic lupus erythematosus, or health problems such as amyloidosis, multiple myeloma and, rarely, carcinoma of the lung and lymphoma. Other patients only have a skin problem.
Researchers have detected tissue-bound immunoglobulin (Ig)-G autoantibodies directed against the NC1 domain of type VII collagen (Col7) in affected patients. Type VII collagen is the major structural component of anchoring fibrils. These attach the basement membrane of the epidermis to the underlying dermis. The loss of anchoring fibrils leads to the formation of blisters just under the epidermis within an area known as the lamina densa.
EBA has several distinct clinical presentations.
Classification of EBA |
Clinical features |
|---|---|
Classical mechanobullous form of EBA |
|
Non-classical/non-mechanobullous form of EBA (inflammatory EBA) |
|
Predominantly mucous membrane form of EBA |
|
Brunsting‐Perry‐type EBA |
|
IgA‐EBA |
|
The following tests should be performed to establish a diagnosis of EBA.
Other tests to confirm the diagnosis are only available in some academic centres. They include [2]:
The International Bullous Diseases Group (IBDG) have proposed by consensus clinical and diagnostic criteria for EBA [1,2]. If a blistering disease is clinically and histologically compatible with EBA, the diagnosis is confirmed if:
EBA may mimic other inflammatory blistering diseases, most often bullous pemphigoid, linear IgA disease, mucous membrane pemphigoid, and bullous systemic lupus erythematosus (which is also associated with anti-Col7 autoantibodies).
The non-inflammatory mechanobullous-type may mimic porphyria cutanea tarda.
In comparison to bullous pemphigoid, EBA [2]:
The primary aim in the treatment of EBA is to protect the skin and stop blister formation, promote healing and prevent complications.
Because EBA is considered an autoimmune disease it is reasonable to use immunosuppressive agents to modify or reduce autoimmune responses and decrease the production of autoantibodies. These may include:
Due to the rarity of the disease, it is difficult to be sure which drug is the most effective.
Other important management strategies include:
EBA is a chronic inflammatory disease that has periods of partial remissions and exacerbations. If treated and cared for properly, patients can expect to live a normal lifespan. Problems that may arise include: