Introduction Demographics Causes Clinical features Complications Diagnosis Differential diagnoses Treatment Outcome
Gardner syndrome is a variant of familial adenomatous polyposis (FAP) that is associated with extra-colonic features. It is an inherited disease that is characterised by gastrointestinal polyps, multiple osteomas (benign bone tumours), and various skin and soft tissue tumours.
Polyps tend to form at puberty with the average age of diagnosis around 25 years of age. Polyps have an almost 100% risk of undergoing malignant transformation resulting in colorectal cancer, therefore timely detection of Gardner syndrome is essential.
The incidence of familial adenomatous polyposis (FAP) is 1 case in 7500 live births. It is due to a congenital autosomal dominant mutation in around 80% of patients. The remaining 20% are spontaneous mutations, with no associated family history.
Although colonic polyps begin to form in puberty, the average age at which Gardner syndrome is diagnosed is 22 years. Osteoma formation usually precedes polyposis. Progression to malignancy is generally observed at age 30–50 years. The average age by which malignancy is detected in patients with Gardner syndrome is 39 years.
Gardner Syndrome is due to mutations on the adenomatous polyposis coli (APC) gene on chromosome 5q22. The gene plays a role in tumour suppression. Gardner syndrome is inherited as an autosomal dominant trait, so an affected person has a 50% chance of passing on the gene to each of their children.
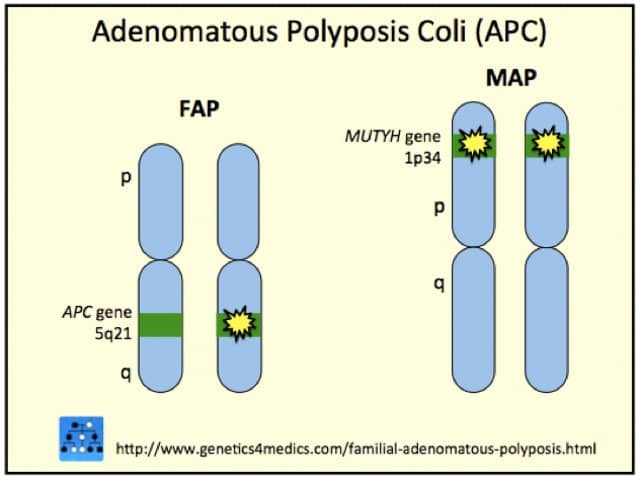
*Image courtesy of Genetics 4 Medics
Clinical features of Gardner syndrome are both cutaneous and non-cutaneous.
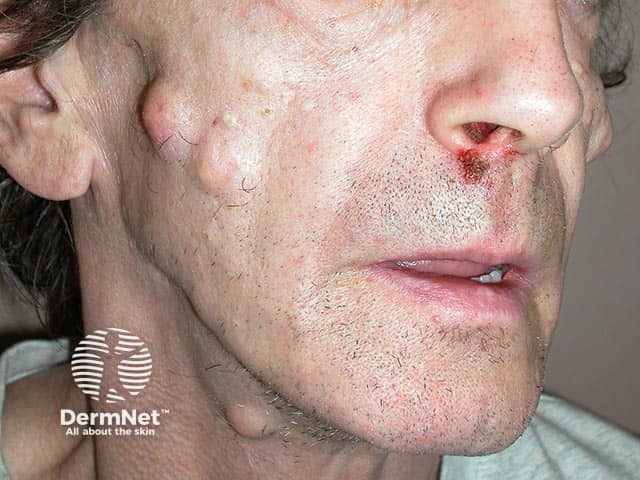
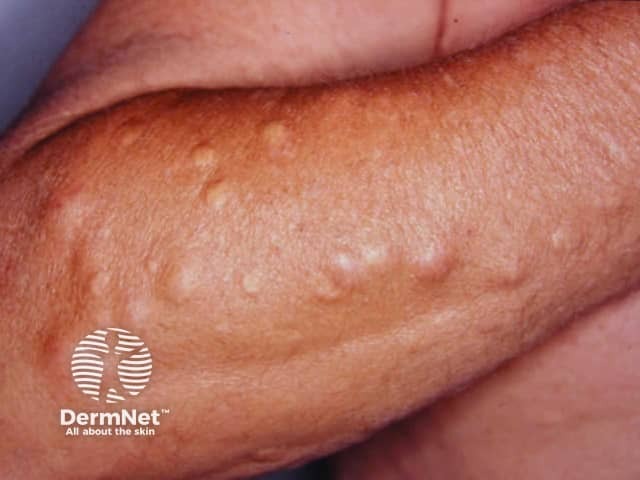
The most noticeable cutaneous feature is the appearance of epidermoid cysts. These can be differentiated from ordinary epidermoid cysts by the following factors:
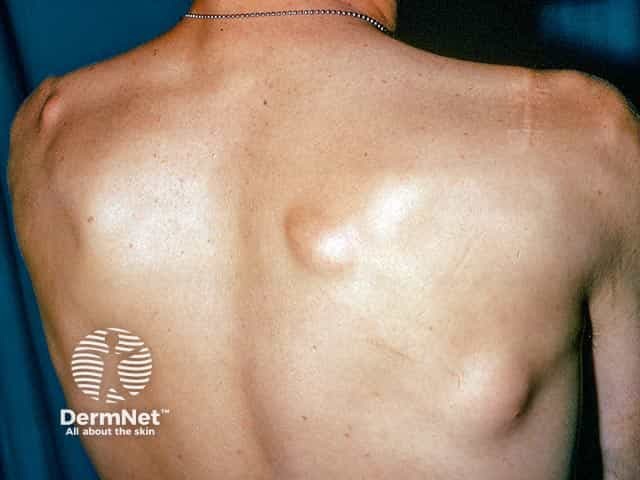
Other cutaneous features include fibromas, lipomas, leiomyomas, and neurofibromas.
Gardner syndrome is diagnosed based on the following features:
Radiological studies are essential for patients and family members with suspected Gardner syndrome.
Differential diagnoses for some features of Gardner syndrome include:
There is no cure for Gardner syndrome. A person with an APC gene mutation will develop colon cancer with almost 100% certainty by around age 40 if surgery is not performed and polyp growth controlled.