Introduction Demographics Causes Clinical features Complications Diagnosis Differential diagnoses Treatment
Steatocystoma multiplex is a rare inherited disorder in which numerous hamartomatous malformations of the pilosebaceous duct junction (hair follicle unit) develop at puberty.
If a single cyst of this type is found, it is called steatocystoma simplex.
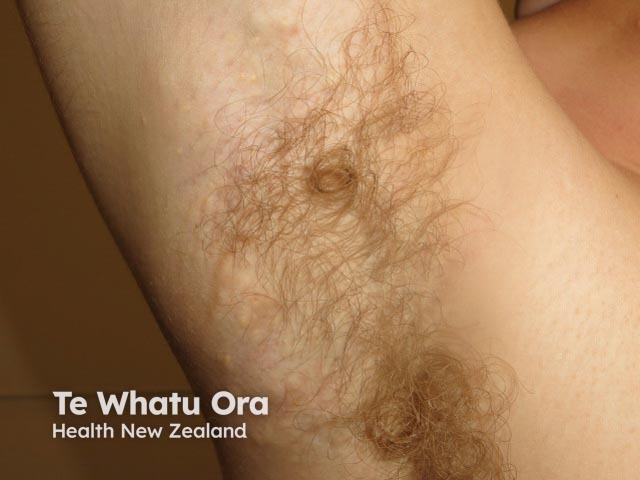
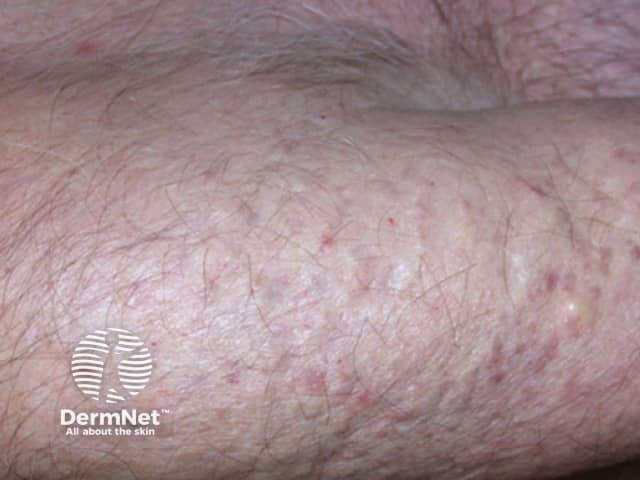
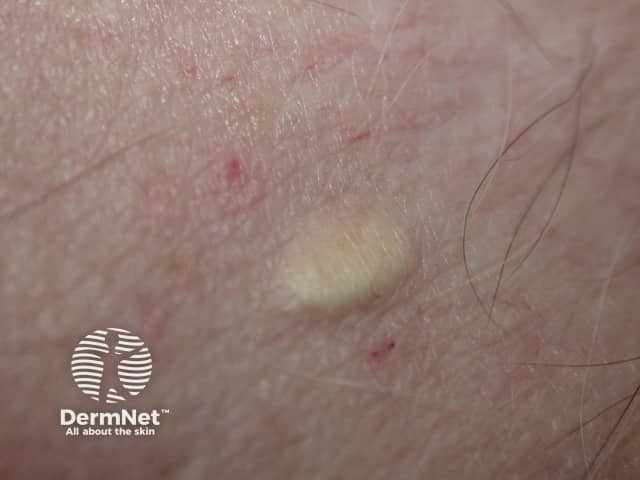
In steatocystoma multiplex, the tendency to develop cysts is inherited in an autosomal dominant fashion, so one parent can be expected to also have steatocystoma multiplex. It may also occur sporadically. Both males and females may be affected.
Familial steatocystoma multiplex is associated with mutations in the keratin 17 gene (K17). This gene is responsible for a keratin protein found in the nail bed, hair follicles and sebaceous glands. Mutations in K17 may also result in one form of pachyonychia congenita, in which cysts, plantar keratoderma and natal teeth are common.
Steatocystomas are thought to come from an abnormal lining of the passageway to the oil glands (sebaceous duct).
The onset at puberty is presumably due to hormonal stimulus of the pilosebaceous unit.
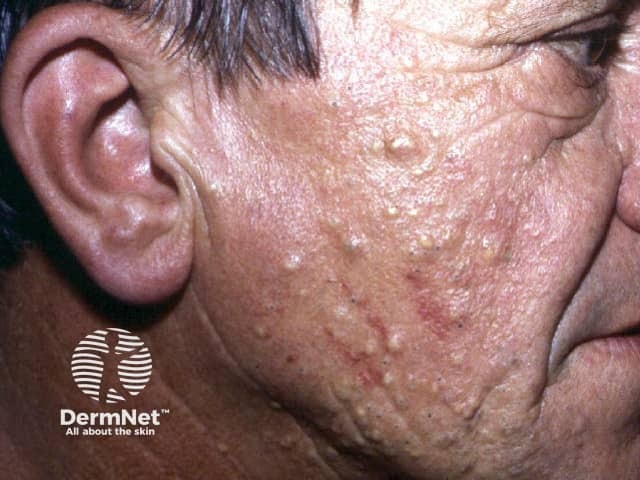
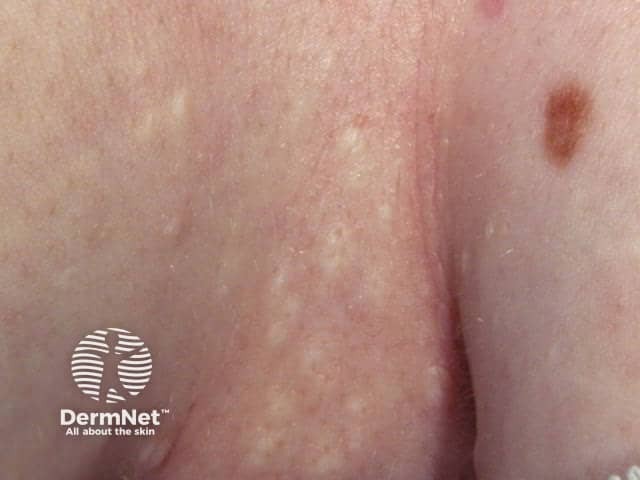
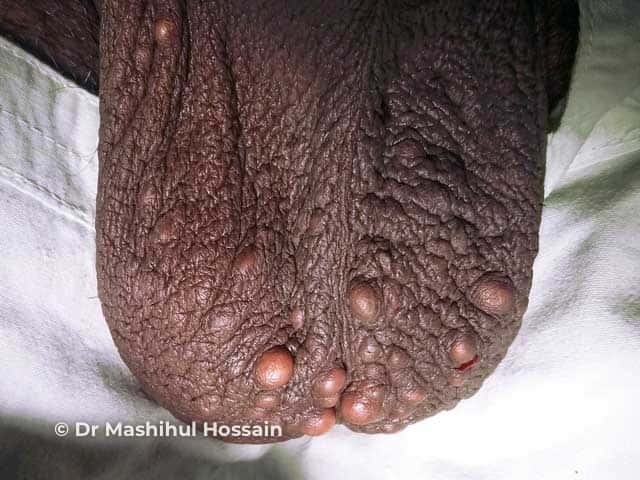
The cysts of steatocystoma multiplex most often arise on the chest and may also occur on the abdomen, upper arms, armpits and face. In some cases cysts may develop all over the body.
The cysts are mostly small (2-20 mm) but they may be several centimetres in diameter. They tend to be soft to firm semi-translucent bumps, and contain an oily, yellow liquid. Sometimes a small central punctum can be identified and they may contain one or more hairs (eruptive vellus hair cysts).
Localised, generalised, facial, acral, and suppurative types of steatocystoma multiplex have been described. Isolated steatocystoma of the vulva and scrotum can develop late in life as a sporadic condition not inherited (see Vulval cysts).
Skin biopsy of a cyst will typically show a thin-walled dermal cyst with a sebaceous gland in the cyst wall and lanugo hair within the cyst lumen.