Introduction Causes Demographics Clinical features Diagnosis Differential diagnoses Outcome
Ataxia-telangiectasia is a rare inherited multisystem disorder that is characterised by:
Ataxia-telangiectasia is also known as Louis-Bar syndrome.
Ataxia-telangiectasia is an autosomal recessive disease in which one ataxia-telangiectasia gene has been inherited from each parent. Carriers of ataxia-telangiectasia with one ataxia-telangiectasia gene and one normal gene do not show any signs of the disease themselves.
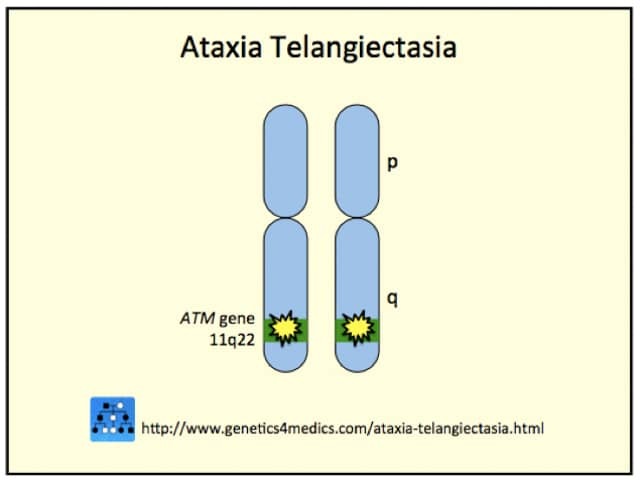
Inactivating mutations of the ATM gene are located on chromosome band 11q22-23. ATM gene codes a protein kinase responsible for double-strand-break DNA repair in the Purkinje cells of the cerebellum and in brain, skin and conjunctival endothelial cells.
A rare variant form of A-T caused by inactivation of the MRE11 gene (11q21) is known as A–T-like disorder.
Ataxia-telangiectasia may occur in any race or sex. Reported rates of incidence range from 1 in 40,000 to 1 in 100,000 births. The incidence of ataxia-telangiectasia is significantly higher when parents are consanguineous, for example in the Bedouin population.
Children with ataxia-telangiectasia appear normal at birth and it is not until they learn to walk at one or two years of age that symptoms become apparent. The severity of the signs and symptoms is variable.
Ataxia results from the progressive degeneration of a part of the brain called the cerebellum. By age 10 or 11 years most patients will need a wheelchair.
Signs and symptoms of progressive neurological impairment include:
The second major clinical manifestation of ataxia-telangiectasia is the development of telangiectases. These usually do not occur until the child is between 3 and 5 years and may not even be apparent until adolescence. The prominent blood vessels first appear around the corners of eyes and then spread to the ears and cheeks.
Patients with ataxia-telangiectasia have a weakened immune system, which makes them vulnerable to recurrent sinus and bronchial infections. They are also at greater risk of getting cancers.
Clinical suspicion of ataxia telangiectasia is backed up by finding:
If a parent is known to have an inactivating ATM gene mutation, prenatal diagnosis can be undertaken.
Other inherited disorders to consider include:
There is no cure for ataxia-telangiectasia. Currently no treatment exists to slow down the progression of the disease. The management of ataxia-telangiectasia is aimed at treating symptoms as they arise, preventing complications and most importantly providing support and education to both patients and their families. This may include physiotherapy, speech therapy, and antibiotics for infection.
Death typically occurs in early or middle adolescence, usually from bronchopulmonary infection, less frequently from malignancy, or from a combination of both. The median age at death in ataxia-telangiectasia is reported to be approximately 20 years. The longest reported survival is 34 years.
Gene therapy holds promise for the future.