Introduction
Demographics
Causes
Clinical features
Complications
Diagnosis
Treatment
Prevention
Outlook
Bullous pemphigoid is the most common form of autoimmune subepidermal blistering disease.
Bullous pemphigoid often presents in people over 80 years of age, and mostly affects people over 50. It can occur in younger adults, but bullous pemphigoid in infants and children is rare.
The most common drugs associated with bullous pemphigoid are the PD1-inhibitor immunotherapies (such as pembrolizumab, nivolumab) used to treat metastatic melanoma and other cancers.
Pemphigoid occurs in 0.3% of cancer patients treated with checkpoint inhibitors, and may develop after 12 months of therapy, or even after they are discontinued. Prodromal itch before blistering and mucosal blisters are common. Drug withdrawal is usually required.
Dipeptidyl peptidase-4 inhibitors (often called 'gliptins'), used to treat diabetes mellitus (such as sitagliptin, saxagliptin, linagliptin and especially, vildagliptin), have also been reported to induce bullous pemphigoid. The median time to onset of bullous pemphigoid is 11 months after initiation of therapy. Unusually, mucosal involvement occurs.
Other cases of bullous pemphigoid have been associated with antibiotics, penicillamine, potassium iodide, frusemide, captopril, gold, penicillin, sulfasalazine, topical fluorouracil, and second-generation antipsychotics (eg, risperidone, olanzapine).
Bullous pemphigoid is the result of an attack on the basement membrane of the epidermis by IgG +/- IgE immunoglobulins (antibodies) and activated T lymphocytes (white blood cells). The target is the protein BP180 (also called Type XVII collagen), or less frequently BP230 (a plakin). These proteins are within the NC16A domain of collagen XVII. They are associated with the hemidesmosomes, structures that ensure the epidermal keratinocyte cells stick to the dermis to make a waterproof seal.
The binding of the autoantibodies to the proteins and release of cytokines from the T cells lead to complement activation, recruitment of neutrophils (acute inflammatory cells) and the release of proteolytic enzymes. These destroy the hemidesmosomes and cause the formation of subepidermal blisters.
The association of neurological diseases with bullous pemphigoid is thought to relate to the presence of collagen XVII in the central nervous system and skin hemidesmosomes.
A rare variant of pemphigoid with IgG antibodies directed against a 200 kDa DEJ protein has been described – there is a strong association with psoriasis (laminin gamma 1).
Bullous pemphigoid causes severe itch and (usually) large, tense bullae (fluid-filled blisters), which rupture forming crusted erosions.
Other variable features include:
Bullous pemphigoid typically involves the flexor aspects of the limbs. It may be localised to one area, or widespread on the trunk and proximal limbs.
Some patients have a diagnosis of bullous pemphigoid made despite not having any bullae (non-bullous pemphigoid). This can affect any site on the body.
All immunobullous diseases may be more severe and recalcitrant in those with skin of colour, and patients tend to have more comorbid conditions, thus complicating treatment.
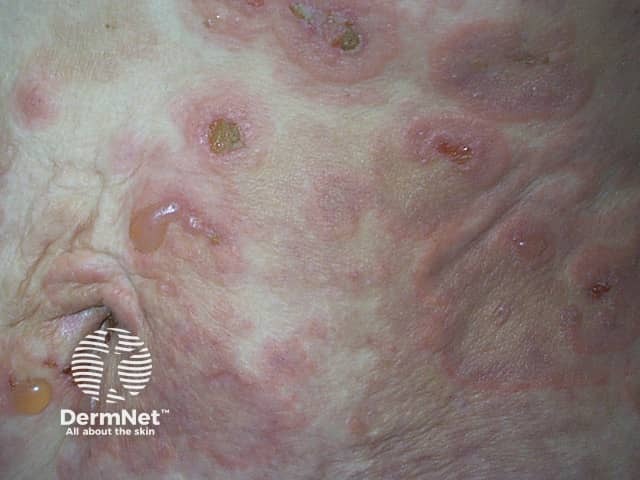
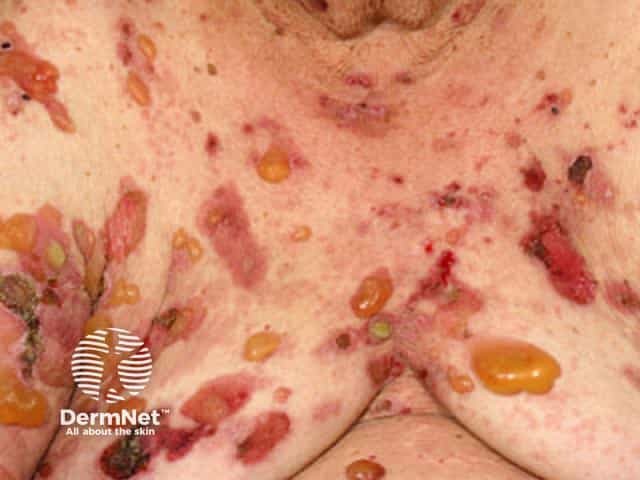
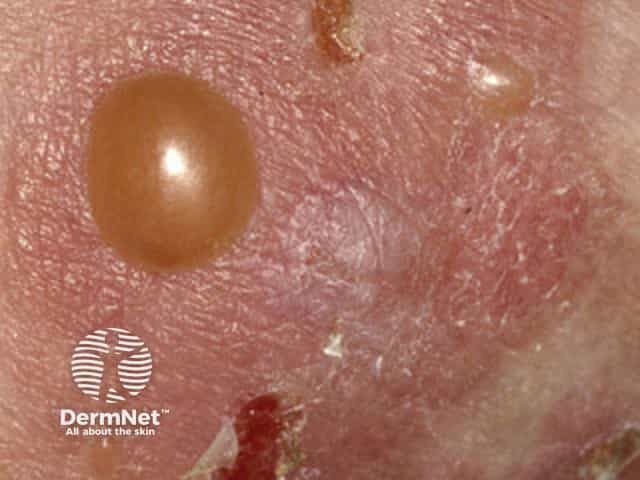
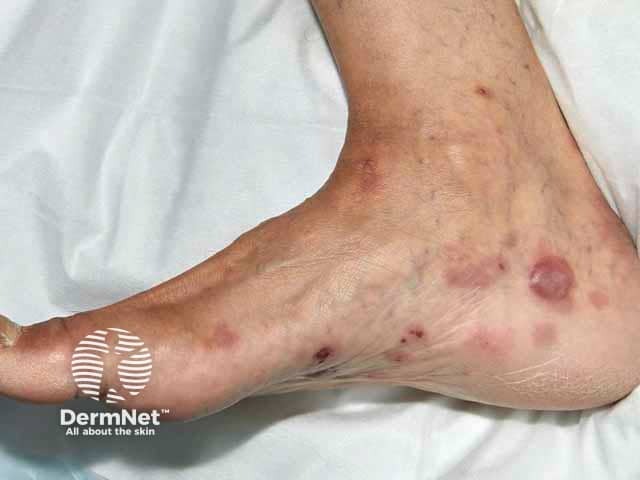
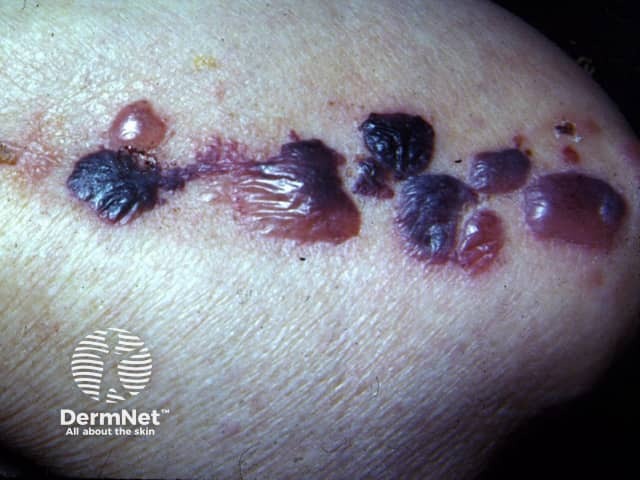
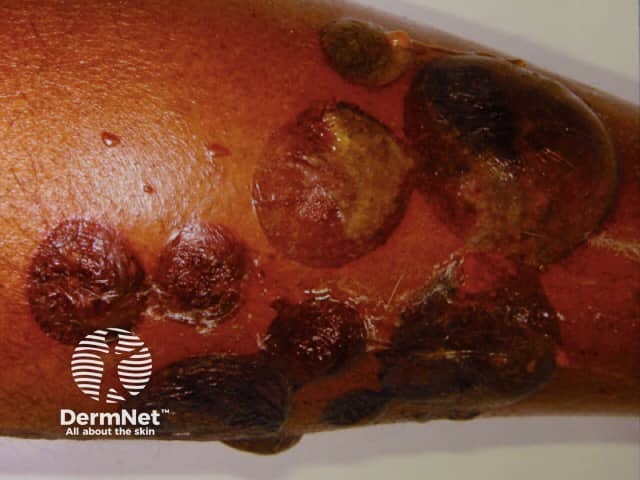
See more images of bullous pemphigoid.
Bullous pemphigoid can be a serious disease, particularly when widespread or resistant to treatment. Morbidity and mortality result from:
When typical bullae are present, the diagnosis is suspected clinically. In most cases, the diagnosis will be confirmed by a skin biopsy of an early blister. The diagnosis can also be made from non-blistered, inflamed skin.
Pathological examination of bullous pemphigoid shows a split under the epidermis. A dermal neutrophilic infiltrate is usual but not always present. Eosinophils may be prominent.
Direct immunofluorescence staining of a skin biopsy taken adjacent to a blister highlights antibodies along the basement membrane that lies between the epidermis and dermis.
Blood tests include an indirect immunofluorescence test for circulating pemphigoid BP180 antibodies.
Immunoblotting.
Other tests will relate to planning and monitoring treatment.
If the pemphigoid is very widespread, hospital admission may be arranged to dress blisters and erosions.
Medical treatment involves:
Most patients with bullous pemphigoid are treated with either ultrapotent topical steroids or steroid tablets, either prednisone or prednisolone at an initial dose of 0.5 mg/kg/day. The dose is adjusted until the blisters have stopped appearing, which usually takes several weeks. The dose of prednisone is then slowly reduced once there are fewer than 3 significant blisters, over many months or years. As systemic steroids have many undesirable side effects, other medications are added to ensure the lowest possible dose (aiming for 5–10 mg prednisone daily). These other medications may include doxycycline or:
People on systemic steroids may also receive additional medicines to alleviate their potential side effects (such as gastritis, hypertension, diabetes mellitus, and osteoporosis).
Monoclonal antibodies targeting IgE (such as omalizumab), IL-17 (such as secukinumab), IL-4, IL-13, IL-5 (mepolixumab), eotaxin-1 (bertilumumab), complement-specific serine protease C1 (sutimlimab), JAK inhibitors, and other mediators are under investigation for the treatment of bullous pemphigoid.
As systemic treatment may be required for bullous pemphigoid for long periods, the extent and severity of the disease should be recorded carefully at baseline and follow-up appointments. The following aspects may be considered.
The Bullous Pemphigoid Disease Area Index (BPDAI) has separate scores for skin and mucous membrane activity.
Blood pressure, body weight, DEXA bone scan, and blood tests are required to monitor therapy, as medications used for bullous pemphigoid may have serious side effects in some patients.
It is not known how to prevent bullous pemphigoid.
Treatment for bullous pemphigoid is usually needed for several years. In many cases, the pemphigoid eventually completely clears up, and the treatment can be stopped. If blisters recur, treatment can be started again.