Introduction - epidermolysis bullosa
Introduction
Demographics
Causes
Clinical features
Diagnosis
Treatment
Outcome
Epidermolysis bullosa (EB) is a group of inherited diseases that are characterised by blistering lesions on the skin and mucous membranes. These may occur anywhere on the body but most commonly appear at sites of friction and minor trauma such as the feet and hands. In some subtypes, blisters may also occur on internal organs, such as the oesophagus, stomach and respiratory tract, without any apparent friction.
Dystrophic epidermolysis bullosa (DEB) is characterised by the site of blister formation in the lamina densa within the basement membrane zone and the upper dermis. It causes generalised blistering of the skin and internal mucous membranes and leads to scar formation.
Dystrophic epidermolysis bullosa is a rare inherited disease. There are two main subtypes: autosomal dominant dystrophic epidermolysis bullosa (DDEB) and autosomal recessive dystrophic epidermolysis bullosa (RDEB). The latter is the more severe form.
Dominant dystrophic epidermolysis bullosa is caused by heterozygous mutation in the type VII collagen gene (COL7A1; 120120 ) on chromosome 3p21.
Recessive dystrophic epidermolysis bullosa is due to homozygous or compound heterozygous mutation in the gene encoding type VII collagen (COL7A1; 120120) on chromosome 3p21. Milder variants of recessive dystrophic EB have been reported with mutations affecting the intron 19 acceptor splicing site of COL7A1.
DEB Subtypes |
Features |
|---|---|
Dominant generalised EB (DEB) |
|
Generalised severe recessive EB (RDEB) |
|
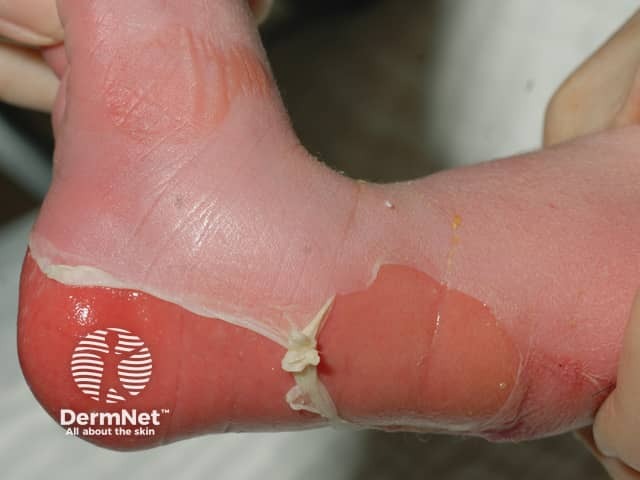
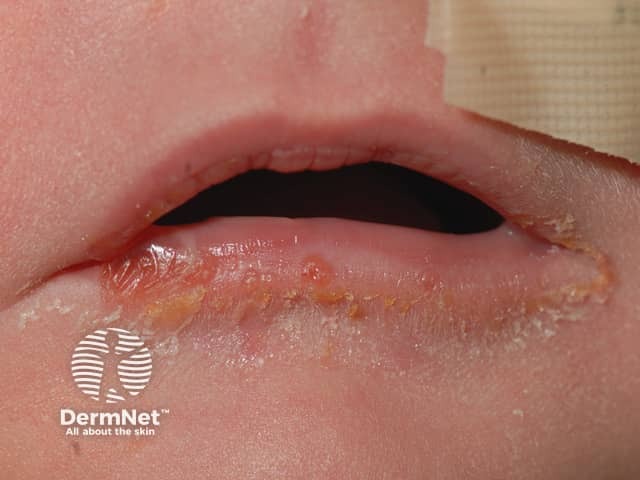
In the dominant subtypes of epidermolysis bullosa, where an informative family tree is known, it is often acceptable for clinical diagnosis to be made by a specialist dermatologist based on the presenting signs.
Squamous cell carcinoma in dystrophic epidermolysis bullosa is diagnosed by its clinical appearance and supported by biopsy.
See treatment of epidermolysis bullosa – general.
Life expectancy is unaffected in dominant dystrophic epidermolysis bullosa. In recessive dystrophic epidermolysis bullosa, life expectancy has significantly improved due to appropriate management and interventions related to complications of the disease including the early detection and treatment of squamous cell carcinoma (SCC).