Introduction
Demographics
Causes
Signs and symptoms
Diagnosis
Treatment
Elastosis perforans serpiginosa (EPS) is a rare skin disorder in which abnormal elastic tissue fibre passes from the papillary dermis (inner layer of skin) to the epidermis (outer layer of skin), described as transepithelial elimination. It presents as a group of small reddish bumps.
Three forms of elastosis perforans serpiginosa have been identified:
Elastosis perforans serpiginosa most commonly appears during early adulthood (20–30 years) but may be seen in early childhood or late in life. It appears to be more common in males than females (ratio 4:1).
Elastosis perforans serpiginosa occurs because the epidermis (the outer layer of the skin) perceives abnormal elastic tissue to be a foreign object, thus responding through an inflammatory attack. The cause of abnormal elastic tissue is unknown but may be from a genetic mutation or external factors such as penicillamine.
Elastosis perforans serpiginosa presents as a cluster of small reddish bumps 2-5 mm in diameter, often grouped in linear, circular or serpiginous (snake-like) patterns. Each lesion may have a central pit, which is sometimes filled by a crusty or scaly plug. Usually, there are no symptoms, but sometimes elastosis perforans serpiginosa is itchy.
Elastosis perforans serpiginosa usually arises on the back of the neck (70%) and less frequently on one or both arms (20%), face (11%), legs (6%) and trunk (3%).
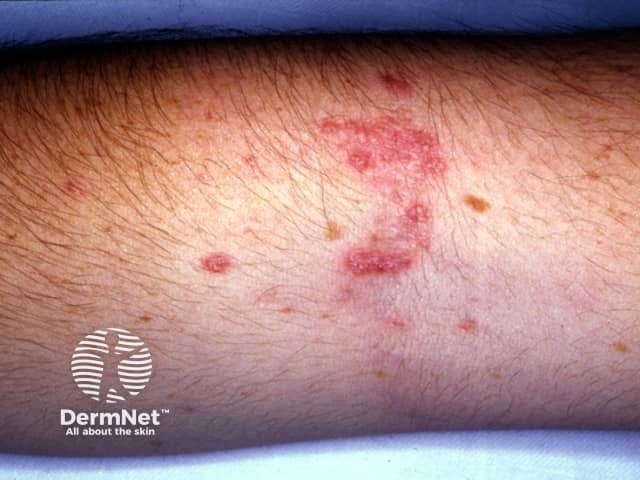
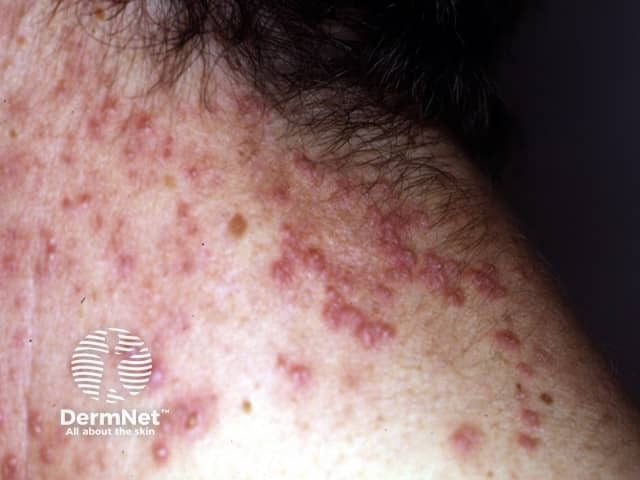
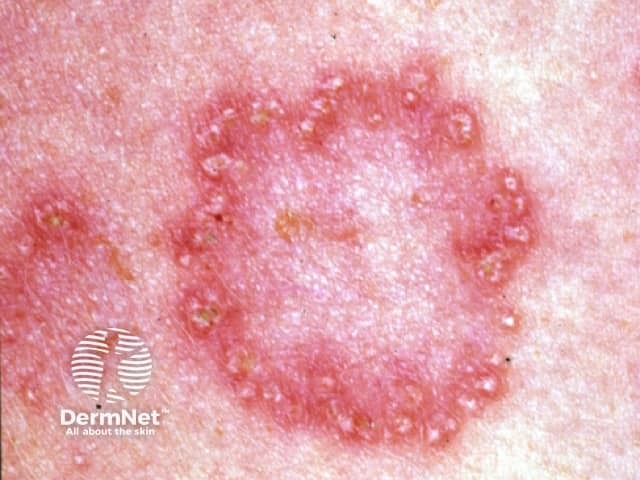
The diagnosis of elastosis perforans serpiginosa is made by its clinical appearance and by the characteristic histology of elastosis perforans serpiginosa seen on skin biopsy, showing transepidermal elimination of elastic tissue (and not elastosis).
There is no cure for elastosis perforans serpiginosa. Usually, it spontaneously resolves without complications after a few years.