Introduction Causes Introduction - IKBKG gene Clinical features Cutaneous features Treatment
Incontinentia pigmenti is a rare genetic condition characterised by skin, eye, teeth and central nervous system (CNS) abnormalities. The characteristic skin lesions of incontinentia pigmenti are present at birth or develop in the first few weeks of life in approximately 90% of patients.
Incontinentia pigmenti is also referred to as ‘Bloch-Sulzberger syndrome’, ‘Bloch-Siemens syndrome’, ‘melanoblastosis cutis linearis’, and ‘pigmented dermatosis-Siemens-Bloch type’.
Incontinentia pigmenti is a dominant X-linked disease. This means that the abnormal incontinentia pigmenti gene is located on one of the X chromosomes, which determine the sex of a child (XY=male; XX=female). Dominant X-linked disease means that a female with only one copy of the abnormal gene will show the disease, even though they have a normal gene on their other X-chromosome. Males who inherit the abnormal gene do not survive, resulting in miscarriage or stillbirth (X-linked dominant, male-lethal syndrome). Rarely incontinentia pigmenti is reported in males with Klinefelter syndrome (XXY syndrome) or as a result of spontaneous mutations.
The incontinentia pigmenti gene is localised on chromosome Xq28. This gene normally codes for the nuclear factor-KB essential modulator protein and is known as the IKBKG gene (formerly known as NEMO or NF-kappaB gene).
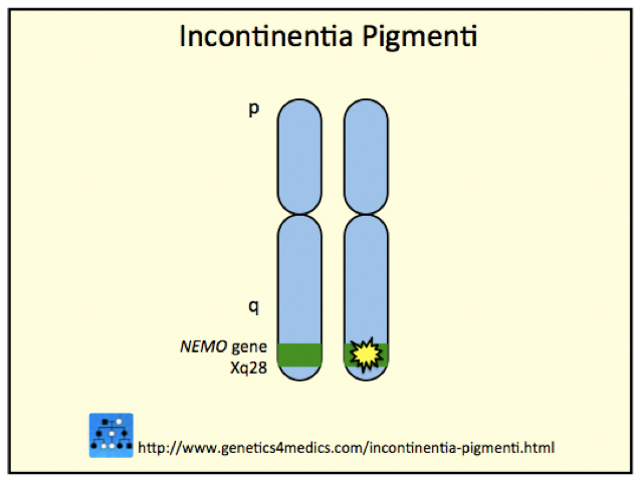
*Image courtesy Genetics 4 Medics
The IKBKG gene is involved in the regulation of the cell’s division and programmed cell death.
Mutations in the IKBKG gene prevent it from working, and cells that have the mutation are more prone to programmed cell death.
Cell death in the skin may present with blisters. These heal as the cells with the mutation die and are replaced by surrounding cells.
Cell death also affects the endothelial cells (cells lining blood vessel walls) in the brain. This causes abnormal vessels to develop, and leakage of proteins from the blood into the brain. This may cause seizures.
Progressive skin rashes are the main clinical feature of the disease. There are four recognised clinical stages but their sequence is irregular, their duration variable and they may overlap.
Stage 1: Vesicular
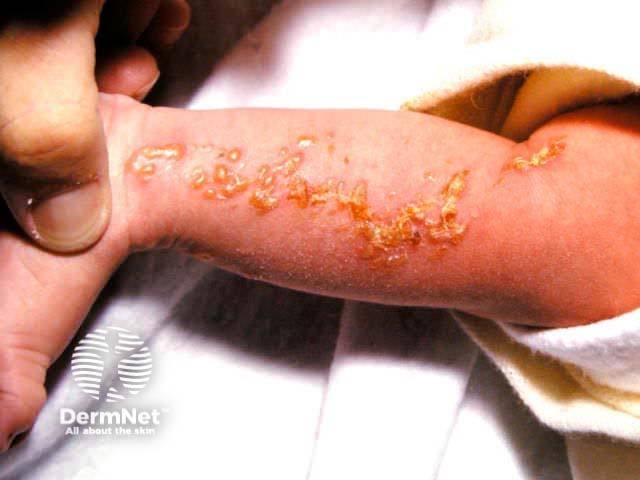
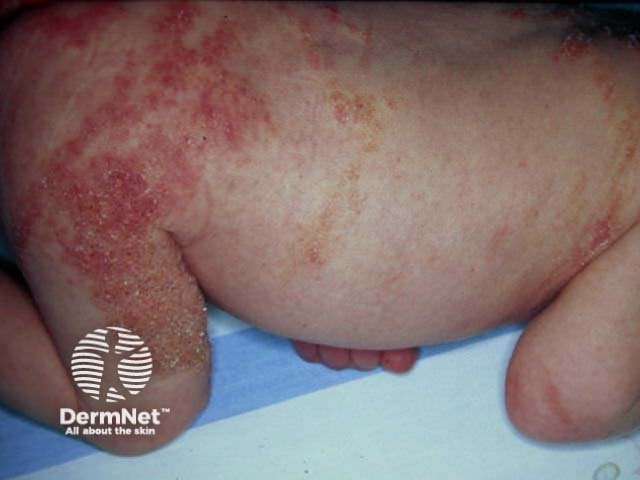
Stage 2: Verrucous
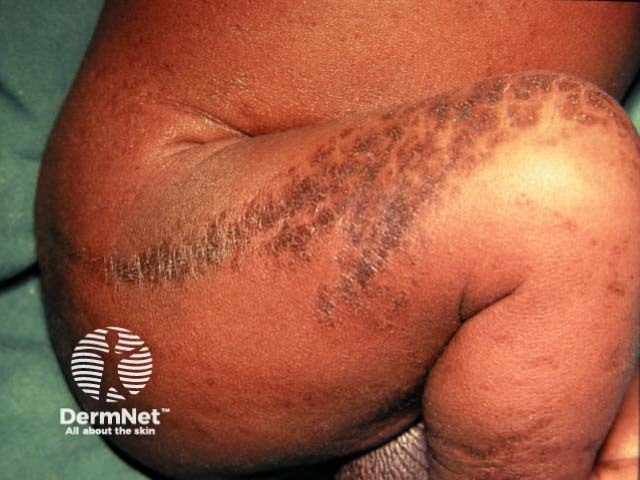
Stage 3: Hyperpigmented
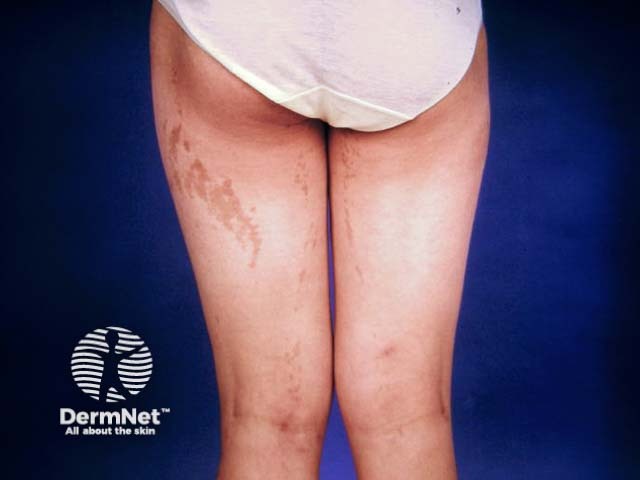
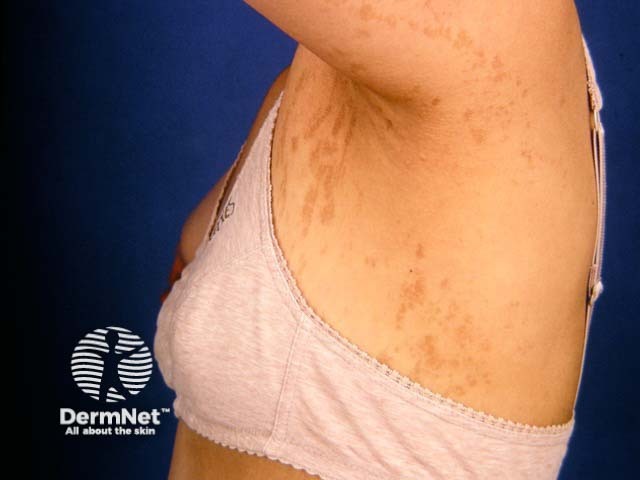
Stage 4: Atrophic/ hypopigmented
Other organs may be affected in various ways in patients with incontinentia pigmenti. These manifestations may not be seen or recognised until infancy or early childhood.
Teeth
Nails
Hair
Eyes
Central nervous system
Other organ systems
There is no specific treatment for incontinentia pigmenti. The main goal is to prevent secondary bacterial infection of skin lesions and to monitor closely the development of related problems. This should include regular dental care and close monitoring by an ophthalmologist for the first few years of life.