What are autoimmune blistering diseases in the eye?
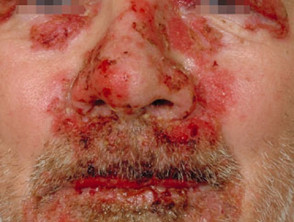
Autoimmune disease (bullous) diseases usually present with blistering and erosions of the skin. They can also involve mucous membranes including the eye, mouth and gastrointestinal system.
Ocular involvement in autoimmune blistering diseases has been reported to occur both before and after the onset of skin lesions [1,2].
Ocular involvement in autoimmune blistering diseases
Who gets autoimmune blistering diseases in the eye?
Eye involvement affects people with autoimmune blistering skin diseases to a varying degree. It is seen most commonly in:
- Mucous membrane pemphigoid
- Epidermolysis bullosa acquisita
- Linear immunoglobulin A bullous disease
- Pemphigus vulgaris
- Paraneoplastic pemphigus.
While ocular involvement in bullous pemphigoid, pemphigus foliaceus, and dermatitis herpetiformis has also been reported, it is much less common.
What causes autoimmune blistering diseases in the eye?
Autoimmune disease arises from a combination of environmental triggers and genetic susceptibility. Ocular involvement is thought to be due to biochemical and ultrastructural similarities common to the skin and cornea [3], which are both embryonically derived from the surface ectoderm [4].
The initial stage of ocular involvement is autoimmune-induced conjunctival inflammation.
- Autoantibodies and complement are deposited in the basement membrane zone or intracellular substance of the conjunctival epithelium [5].
- Activated inflammatory cells release cytokines.
- Remodelling of the extracellular matrix (the layer of molecules that give structural support to the skin) leads to progressive fibrosis of the eyelid, conjunctiva or cornea [6].
Complications vary in frequency and severity among the different types of autoimmune blistering diseases, but all have the potential for serious morbidity and can threaten sight.
What are the symptoms of autoimmune blistering diseases in the eye?
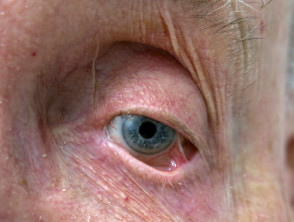
Blistering diseases mainly affect the eyelids, conjunctiva, and cornea. The disease may initially arise in one eye but generally progresses to affect both eyes. Patients may experience:
- Burning/soreness
- Crusting of eyelids
- Decreased vision
- Dry eye
- Foreign body sensation
- Increased tears
- Irritation
- Mucous discharge
- Ocular pain
- Photophobia (light sensitivity)
- Red eye
- Tearing.
Patients may be asymptomatic even with advanced ocular signs [6].
What are the clinical signs of autoimmune blistering diseases in the eye?
The clinical signs of autoimmune blistering diseases affecting the eye are due to progressive chronic cicatrising (scarring) conjunctivitis.
- Conjunctival goblet cells are destroyed.
- Lacrimal gland ductules and meibomian gland orifices are obstructed, reducing tear production and resulting in keratoconjunctivitis sicca (dry eye) [7].
- Deficiency and destabilisation of the tear film make the surface of the eye vulnerable to external damage [8].
Other common signs in early disease include:
- Conjunctival hyperaemia (dilated conjunctival blood vessels)
- Subconjunctival bullae
- Fibrosis of the subepithelial tissue causing shortening of the fornices (loose folds of conjunctiva between the inside of the eyelids and the eyeball)
- Symblepharon formation (adhesion of the eyelid to the eyeball).
- Corneal and conjunctival keratinisation (formation of thickened epithelium).
Common eyelid complications include:
- Ankyloblepharon (eyelids have adhered together)
- Blepharitis (inflammation of eyelid margin)
- Distichiasis (the presence of a double row of eyelashes)
- Ectropion (the eyelid is turned inside out)
- Entropion (the eyelid is turned inward)
- Madarosis (loss of eyelashes)
- Trichiasis (the eyelashes are turned towards the eyeball).
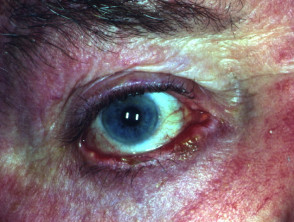
Epithelial defects of the cornea may progress to ulceration, scarring, and secondary bacterial infection. End-stage ocular involvement is characterised by:
- Complete destruction of the conjunctival fornices (conjunctival shrinkage)
- Corneal neovascularisation (growth of new blood vessels into the cornea)
- Opacification of the ocular surface (clouding of the cornea)
- A decrease in vision or complete blindness.
The clinical findings and complications of ocular involvement often overlap between the different types of autoimmune blistering skin diseases. Spontaneous remissions are rare.
How are autoimmune blistering diseases in the eye diagnosed?
Ocular involvement should be suspected in patients with autoimmune blistering diseases. Early detection and management are vital to prevent sequelae such as irreversible vision loss.
Evaluation by an ophthalmologist is highly recommended as part of routine multidisciplinary care. This will usually involve a thorough history, and:
- Measurement of visual acuity
- Slit-lamp examination
- Tear break-up time
- Schirmer test for dry eye
- The adding of fluorescein and lissamine dyes to the eyes to detect corneal defects.
Clinical diagnosis is usually confirmed by immunopathology.
- Direct immunofluorescence on a conjunctival biopsy may demonstrate immunoglobulin or complement deposition at the basement membrane zone [9].
- Indirect immunofluorescence of serum (a blood test) may reveal circulating IgG autoantibodies.
- Other tests include immunoblotting (blood tests) and enzyme-linked immunosorbent assay (ELISA).
What are the differential diagnoses for autoimmune blistering diseases in the eye?
Diseases that may have similar ocular manifestations to autoimmune bullous diseases include:
- Ocular trauma/irradiation
- Ocular rosacea
- Atopic keratoconjunctivitis
- Chemical burns
- Adverse reaction to systemic medications (eg, practolol and penicillamine)
- Adverse reaction to eye drops (eg, echothiopate iodide, epinephrine, and pilocarpine)
- Graft versus host disease
- Inherited epidermolysis bullosa
- Lichen planus
- Sarcoidosis
- Sjögren syndrome
- Stevens–Johnson syndrome/toxic epidermolysis necrolysis.
These can be differentiated from ocular involvement in autoimmune bullous diseases by a careful history, a complete review of the patient's system, an examination and immunopathological investigations [9].
What is the treatment for autoimmune blistering diseases in the eye?
Treatment of ocular autoimmune blistering diseases is individualised, based on age, disease severity, and other sites of involvement [7]. The primary aim of treatment is to control ocular inflammation and prevent complications or progression to irreversible scarring and blindness. Once controlled, the goal is to taper doses of medication until a minimal maintenance dose or drug-free remission can be attained. Close collaboration between the treating dermatologist and ophthalmologist is critical.
Conservative management
- Frequent lubrication using artificial tears or ointment can alleviate symptoms of dry eye.
- Regular lid hygiene using dilute bicarbonate solution or a proprietary preparation applied via a clean finger or cotton bud may improve symptoms of blepharoconjunctivitis.
- Warm compresses to closed eyelids for 5–10 minutes daily can improve the flow of meibomian gland secretions.
- Contact lens use should be avoided to prevent eye irritation. A special bandage contact lens may be employed to provide protection from exposure and trichiasis while awaiting surgical management [10].
- Topical corticosteroid eye drops may contain dexamethasone, fluocinonide, triamcinolone acetonide or clobetasol propionate. They may be used in early stage disease or acute flare-ups.
- Topical retinoids may reduce keratinisation of the conjunctival lid margin but should be used only for a few weeks.
- Ciclosporin eye drops have been shown to improve tear production in dry eye.
Medical management
Systemic corticosteroids (eg, oral prednisone or prednisolone) are the first-line therapy for treating ocular involvement in autoimmune blistering skin diseases [11]. Due to the significant toxicity of corticosteroids at the high doses required, other immune-suppressive/regulatory therapies are added, such as:
- Azathioprine
- Colchicine
- Cyclophosphamide [12]
- Dapsone
- Methotrexate
- Mycophenolate mofetil
- Sulfasalazine
- Intravenous immunoglobulin
- Biological agents (eg, etanercept, rituximab, daclizumab, alemtuzumab).
Tetracyclines are used to treat secondary corneal infections and blepharoconjunctivitis.
Treatments are continued long term and may require regular monitoring, due to potential side effects.
Surgical intervention
Surgery is rarely indicated in early disease and can induce exacerbation or progression when performed on inflamed eyes [13]. Surgical intervention should be delayed until the eyes are no longer inflamed and progression has stabilised.
- Punctal plugs (temporary) or surgical punctal occlusion (permanent) may be used in a severe dry eye.
- Cryotherapy, laser thermal ablation or electrolysis are used to prevent regrowth of eyelashes in contact with the eyeball.
- Repositioning of the eyelid may be warranted in entropion, ectropion, and lagophthalmos (an inability to close the eyelids completely).
- Amniotic membrane grafting may be necessary to repair extensive scarring affecting the conjunctiva.
- Penetrating keratoplasty (plastic surgery on the cornea, especially corneal grafting) and limbal stem cell transplantation can be used to treat corneal perforation.
- Keratoprostheses (artificial cornea) may be recommended if corneal grafting has failed.