What is systemic sclerosis?
Systemic sclerosis (SSc) is an autoimmune inflammatory condition. It results in potentially widespread fibrosis and vascular abnormalities, which can affect the skin, lungs, gastrointestinal tract, heart and kidneys. The skin becomes thickened and hard (sclerotic).
Systemic sclerosis has been subdivided into two main subtypes, according to the distribution of skin involvement.
- Diffuse cutaneous systemic sclerosis (dcSSc)
- Limited cutaneous systemic sclerosis (lcSSc)
Limited cutaneous systemic sclerosis was previously referred to as CREST syndrome to denote key features:
- Calcinosis
- Raynaud phenomenon
- (o)Esophageal dysmotility
- Sclerodactyly
- Telangiectases.
The general term ‘scleroderma’ is often used for both morphoea (localised scleroderma) and systemic sclerosis (systemic scleroderma). Distinguishing these two conditions is very important, as they vary greatly and require different treatment.
Key features of systemic sclerosis
- Skin thickening of the fingers and toes (sclerodactyly)
- Specific autoantibodies in the blood (anti-Scl70 or anti-centromere antibody and others)
- Abnormal nail fold capillaries
- Internal organ fibrosis and/or vascular damage (involving the lungs, heart, gastrointestinal tract and/or kidneys)
Key features of systemic sclerosis
Who gets systemic sclerosis?
Systemic sclerosis is rare, with prevalence varying from 30–500 cases per million.
- It is up to 5 times more common in females compared to males.
- All races and ethnicities may be affected, but rates appear to be slightly higher in some Native Americans and black-skinned races and lower in those of Asian background.
- The peak age of onset of SSc varies between approximately 35 and 55 years. Juvenile onset occurs but is rare compared to adult-onset disease.
What causes systemic sclerosis?
Systemic sclerosis is an autoimmune condition characterised by inflammation, fibrosis and vasculopathy.
The precise underlying mechanisms are complex and remain largely unknown. Genetic susceptibility plus a triggering event result in a cascade of innate and adaptive immunoinflammatory responses.
Genetic susceptibility
- First degree relatives of affected individuals may be at 10–16 fold increased risk of developing systemic sclerosis, compared with those with no family history of the disease.
- Studies have identified genetic loci associated with systemic sclerosis.
- Clinical subtypes map to particular genetic subsets.
- Differences in gene expression occur in fibroblasts, immune (T and B), endothelial, smooth muscle and epithelial cells.
A triggering event
Systemic sclerosis has been associated with injury, exposure to silica, vinyl chloride monomer, chlorinated solvents, trichloroethylene, welding fumes, aromatic solvents, ketones, bleomycin and possibly other drugs (vitamin K, cocaine, penicillamine, appetite suppressants and some chemotherapeutic agents).
Immune pathways
A number of pathways are likely involved in the pathogenesis of systemic sclerosis, including cytokines that injure blood vessels, growth factors that stimulate collagen production, integrin signalling, morphogen pathways, co-stimulatory pathways and more.
How is systemic sclerosis classified?
Diffuse cutaneous systemic sclerosis
Two-thirds of patients with systemic sclerosis have dcSSc: skin involvement is widespread and includes proximal limbs. DcSSC is often rapidly progressive, with significant internal organ involvement.
Limited cutaneous systemic sclerosis
One-third of patients with systemic sclerosis have lcSSc: sclerosis is limited to the digits, distal limbs (not spreading more proximal than the elbows or knees) and face. LcSSc progresses more slowly than dcSSc and with less internal organ involvement except there is a risk of pulmonary artery hypertension, especially later in the disease course.
Overlap syndrome
Up to 20% of patients with systemic sclerosis have an overlap syndrome with another connective tissue disease and develop arthritis, lupus or myositis.
Systemic sclerosis sine scleroderma
Systemic sclerosis sine scleroderma is a rare subtype without skin sclerosis. These patients have SSc-related internal organ manifestations, Raynaud phenomenon and SSc-specific auto-antibodies.
Serological and genetic classification
Different autoantibody profiles are associated with particular clinical features and prognosis, particularly the pattern of antinuclear antibody (ANA) reactivity. Genetic associations in systemic sclerosis can also be mapped to particular ANA subtypes.
Centromere (kinetochore) ANA pattern
- Limited cutaneous systemic sclerosis
- Pulmonary artery hypertension
- Digital ulcers
- Calcinosis
- Associated genes: HLA-DQB1, TNF-863A, NOTCH4
Pm-Scl (nucleolar) ANA pattern
- Myositis overlap
U1RNP (speckled) ANA pattern
- Overlap with features of other connective tissue diseases
- Associated gene: HLA-DQB1
Th-To (nucleolar) ANA pattern
- Limited cutaneous systemic sclerosis
- Pulmonary artery hypertension
- Interstitial lung disease
- Poor prognosis
Topoisomerase-1 / Scl-70 (speckled) ANA pattern
- Diffuse cutaneous systemic sclerosis
- Interstitial lung disease
- Cardiac scleroderma
- Associated genes: HLA-DPA1, HLA-DRB1
RNA polymerase (P) III (fine speckled, nucleolar) ANA pattern
- Diffuse cutaneous systemic sclerosis
- Scleroderma renal crisis
- Malignancy
- Pulmonary artery hypertension
- Associated genes: HLA-DRB1, HLA-DRB3, HLA-DRB4, EDNRA
Fibrillarin / U3RNP (nucleolar)
- Afro-Caribbean association
- Pulmonary artery hypertension
- Myositis
- Cardiac scleroderma
- Gastrointestinal involvement
- Associated gene: HLA-DQB1
What are the clinical features of systemic sclerosis?
The clinical features of systemic sclerosis are related to underlying vascular, inflammatory and fibrotic disease. Constitutional symptoms are common, such as fatigue, arthralgia and myalgia.
Cutaneous features of systemic sclerosis
Skin sclerosis
- The extent of skin fibrosis defines 'diffuse' versus 'limited' systemic sclerosis.
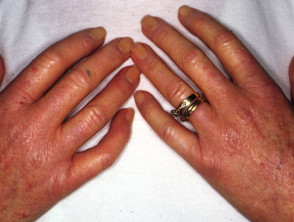
- Sclerodactyly: thickening and tightness of the skin of the fingers (or toes). Can be spindle-shaped.
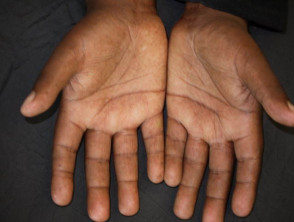
Hands
- Puffy fingers; early inflammatory phase of the disease
- Raynaud phenomenon
- Abnormal nail fold capillaries
- Palmar erythema affecting thenar/hypothenar eminence
- Smaller fragile nails with ragged cuticles
- Digital pitted scars
- Digital ulcers
- Ulceration can lead to dry gangrene and eventual loss of the tips of the fingers (like frostbite).
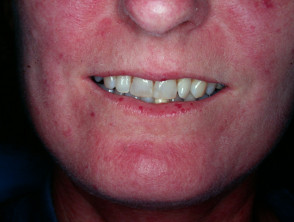
Face
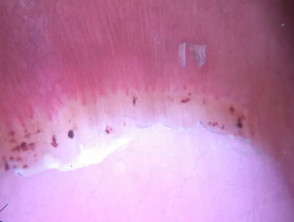
- Matt telangiectases on face, chest, palms
- Peri-oral furrowing (fat loss)
- Microstomia (limited oral aperture defined as interlabial distance < 4.5 cm)
- Beaked nose
Other
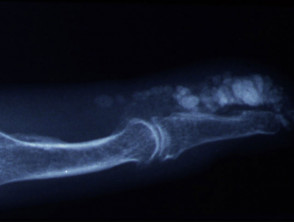
- Calcinosis affecting digits, extensor surfaces of limbs. Skin can breakdown and discharge chalky material (calcium)
- Salt and pepper dyspigmentation (hyperpigmentation and vitiligo-like depigmentation)
- Pruritus
Rarer cutaneous features
- Morphoea
- Most frequently plaque, nodular or linear
- More common in limited cutaneous systemic sclerosis
- Panniculitis
Cutaneous features of systemic sclerosis
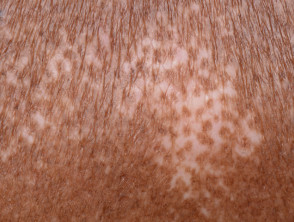
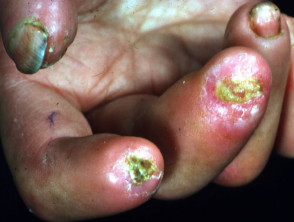
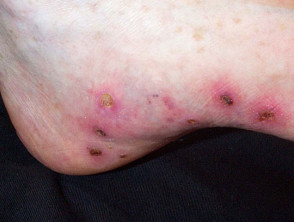
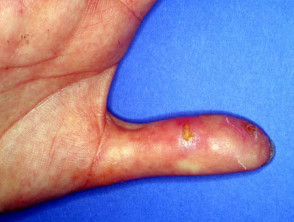
Gastrointestinal symptoms
Upper GIT
- Gastroesophageal reflux/heartburn
- Dyspepsia
- Dysphagia
- Early satiety
- Micro-aspiration (accelerates lung disease)
Lower GIT
- Bloating, distention
- Nausea, vomiting
- Pain
- Alternating diarrhoea/constipation
- Incontinence (anorectal sphincter dysfunction)
Cardiopulmonary symptoms
- Interstitial lung disease
- Pulmonary artery hypertension
- Cardiac scleroderma (cardiomyopathy, conductive)
- Shortness of breath
- Decreased exercise tolerance
- Chest pain
- Palpitations
Renal disease
Scleroderma renal crisis:
- Proteinuria
- High blood pressure
- Renal failure.
Other symptoms
- Fatigue
- Sicca symptoms (dry eyes, dry mouth) and Sjogren syndrome
- Musculoskeletal symptoms: friction rubs over the joints and tendons, particularly the knees; joint pain, muscle pain, weakness and limited movement resulting in contractures
- Ocular symptoms: tight eyelids, reduced tear secretion, retinopathy
Paraneoplastic systemic sclerosis
Malignancy in association with systemic sclerosis is rare.
- Anti-RNAP-III antibodies are found in up to 15% of patients with paraneoplastic systemic sclerosis.
- Patients are usually older than 65 years at presentation.
- Skin involvement is atypical
- The disease tends to be treatment-resistant.
- Associated malignancies include breast, haematological and gastrointestinal cancers.
How is systemic sclerosis diagnosed?
The diagnosis of systemic sclerosis is confirmed when key features are present.
- Sclerodactyly
- Abnormal nail folds capillaries on capillaroscopy/dermatoscopy
- Specific autoantibodies (especially anti-Scl70 or anti-centromere antibody)
- Internal organ fibrosis and vascular damage
Investigations may include:
- Other blood tests: anaemia, raised sedimentation rate (ESR) and C-reactive protein (CRP), positive Rheumatoid factor, increased gamma globulins (hypergammaglobulinaemia), and abnormal coagulation tests may be present.
- Skin biopsy: excessive ground substance and odd-looking endothelial cells in the dermis; and later, deposits of collagen. The epidermis is usually atrophic (thinned).
- Trichoscopy of the forehead using a dermatoscope
- Pulmonary function tests
- High-resolution CT scan
- Echocardiogram
- Right heart catheter
- ECG
- Cardiac MRI
- Barium swallow, manometry
- Endoscopy with gastrointestinal biopsy.
The joint American College of Rheumatology (ACR) and European League against Rheumatism (EULAR) classification criteria (2013) are utilised to diagnose SSc. A score of 9 or more confirms the diagnosis.
- Skin thickening of the fingers of both hands extending proximal to metacarpal phalangeal joint: (score 9)
- Skin thickening of the fingers only: puffy fingers (2); sclerodactyly (4)
- Fingertip lesions: digital tip ulcers (2); fingertip pitted scars (3)
- Telangiectases (2)
- Abnormal nail fold capillaries (2)
- Pulmonary disease: pulmonary artery hypertension (2); interstitial lung disease (2)
- Raynaud phenomenon (3)
- SSc-specific autoantibodies ACA (3), Anti-scl70 (3), Anti-RNA polymerase III 3() (maximum score 3)
How is systemic sclerosis monitored?
Monitoring of progress and treatment response is vital in systemic sclerosis.
Skin
The skin is usually monitored clinically using the modified Rodnan Skin Score (mRSS), which gives an indication of the extent and severity of cutaneous sclerosis, which also reflects the severity and risk of internal organ involvement.
- A score of 0 (normal skin) to 3 (skin not able to be pinched) is assigned for skin tightness for 17 body sites.
- The cumulative score is calculated out of a total of 51; severe skin involvement is defined by a score of > 20.
- A high mRSS is an independent risk factor for a poorer overall outcome.
- Other less readily available instruments can be used to monitor skin hardness, such as serial durometer measurements or more complex measure via cutometer, ultrasound or other measures.
Interlabial distance (mouth opening) can also be measured
- Microstomia is defined as an inter-labial distance of less than 4.5 cm.
Internal organs
Routine annual screening for interstitial lung disease and pulmonary artery hypertension should include:
- Pulmonary function tests
- Transthoracic echocardiogram
- ECG
The DETECT score is a screening tool for pulmonary artery hypertension that uses pulmonary function tests (FVC and DLCO), serum urate, serum NTproBNP and other features to provide a predictive score. A right heart catheter may be indicated in some.
- High-resolution CT scan may be used to characterise interstitial lung disease.
- Blood pressure and renal function monitor renal disease.
- Other organ monitoring is performed on an individualised basis and can include cardiac MRI, oesophageal manometry and gastrointestinal endoscopy.
What is the treatment of systemic sclerosis?
Treatments help with symptoms and may modify the disease outcome, especially early in the disease course. They focus on suppressing inflammation and dilating abnormal / constricted blood vessels. Some newer treatments target specific immunological pathways and signalling molecules.
General advice
- It is absolutely essential for smokers to stop smoking.
- Avoid vasoconstrictive drugs, such as decongestants, amphetamines, ergotamine.
Fatigue, weakness and generalised musculoskeletal symptoms can be debilitating.
- Hydroxychloroquine may help these constitutional symptoms.
- Gentle, controlled exercise may also be of benefit.
- Simple or more complex analgesia may be required.
Anti-fibrotic therapies
Topical therapies
- Warm wax baths (hands)
- Tacrolimus ointment
- Potent corticosteroids
- Calcipotriol
Physical therapies
- Phototherapy, especially UVA1 or PUVA
Systemic therapies
- Mycophenolate mofetil
- Methotrexate
- Cyclophosphamide
- Corticosteroids (with caution)
- Rituximab (B-cell depletion therapy) – may be useful for lung and skin
- Nintedanib (tyrosine kinase inhibitors) — now approved for SSc associated Interstitial Lung Disease in the US and Europe; no proven impact on skin sclerosis
- Tocilizumab (anti-interleukin (IL) 6 therapy) — shows a trend towards improvement for lung and skin in early progressive and inflammatory diffuse cutaneous SSc
- Abatacept (anti-cytotoxic T-cell lymphocyte-associated inhibitor [CTLA] 4 therapy — shows a possible trend towards skin improvement in inflammatory disease )
- Autologous stem cell transplant — for severe and acute progressive disease in selected patients seen within specialist centres
- Intravenous immunoglobulin — may be useful for skin and gastrointestinal tract
- Tofacitinib – well-tolerated, prevents bleomycin-induced skin fibrosis in a mouse model; a small study showed no clinical improvement in skin sclerosis, but larger studies are needed
Vasodilation
- Endothelin 1 antagonists (bosentan)
- Phosphodiesterase-5 inhibitors (tadalafil)
- Guanylate cyclase agonists (riociguat)
- Prostacyclin agonists (epoprostenol; selexipag)
Treatment of skin manifestations
General measures such as keeping warm, stretching exercises for joints to reduce the risk of worsening contractures and microstomia, and specifically-related physiotherapy can all be beneficial.
- Avoid triggers (smoking cessation, protect from cold)
- General measures; double-lined gloves, exothermic hand/feet warmers
- Natural therapies (unproven)
-
- Vitamin C, Vitamin E
- Gamolenic acid
- Medical therapies
- Fluoxetine
- Losartan
- Diltiazem, nifedipine
- Sildenafil, tadalafil
- Glyceryl trinitrate patches
- Prostaglandin
- Botulinum toxin
Digital ulcers
- Ulcer prevention
- Emollients
- Avoid smoking, cold (double-lined gloves), trauma
- Treat underlying Raynaud’s
- Intervene early
- Ulcer treatment
- Localised dressings; protect and keep moist areas of impending ulceration and ulcerated areas
- Systemic vasodilators (see above)
- Combination therapy often advocated and most effective, for example with calcium channel blocker, phosphodiesterase-5 inhibitor (eg, sildenafiil) +/- prostanoids (Iloprost®) +/- endothelin receptor antagonist (eg. Bosentan). Combination therapy often advocated and most effective, for example with calcium channel blocker, phosphodiesterase 5 inhibitor (eg. Sildenafiil) +/- prostanoids (Iloprost®) +/- endothelin receptor antagonist (eg, bosentan).
- Resveratrol is reported to be useful in SSc vasculopathy; this is found in grapes, blueberries, raspberries, and mulberries.
- Surgery
- Amputation
- Botulinum toxin injections
- Digital sympathectomy
- Prevention of ulcer complications
- Pain management
- Infection — 10% infection rate per year
- Cosmetic camouflage (green-tinted makeup)
- Vascular laser or intense pulsed light therapy
Cutaneous calcinosis in SSc is notoriously challenging to treat and controlled trials are lacking. It is difficult to manage, and there is poor evidence for therapies listed below.
- Medical therapies
- Tetracycline antibiotics (6 to 12-week courses)
- Diltiazem
- Bisphosphonates
- Sodium thiosulfate (systemic, intralesional injections, or topical [25%])
- Cinacalcet
- Physical interventions
- Surgical excision
- Curettage and cautery
- Dental drill removal
- Ablative laser
- Extracorporeal shock wave lithotripsy
Pruritus
Pruritus affects up to 43% of those with SSc. Ultimately, pruritus is a sign of active disease and so the SSc itself needs to be treated. Pruritus has been linked to more severe skin and gastrointestinal tract involvement. However, symptom management may also be needed and can include:
- Emollient, soap substitutes
- Topical corticosteroids
- Antihistamines
- Montelukast
- Photochemotherapy (PUVA)
- Low-dose naltrexone
- Neuroactive antipruritics such as gabapentin or doxepin
- Systemic glucocorticoids (with caution)
Facial fat loss
Treatment of internal organ manifestations
Interstitial lung disease
- Antifibrotic, immunosuppressive therapies (see above)
Pulmonary artery hypertension
- Vasodilatory therapies (see above)
Renal disease
- Monitor blood pressure regularly
- Avoid corticosteroids (especially if RNAPIII +ve)
- Angiotensin-converting enzyme inhibitors
Gastrointestinal tract
- Upper GIT: gastro-oesophageal reflux, dyspepsia, dysphagia, aspiration
- Protein pump inhibitors, such as omeprazole
- Alginates (such as Gaviscon®); these are as effective as prokinetics in proton pump inhibitor-resistant gastro-oesophageal reflux disease symptoms in SSc
- Treat Helicobacter pylori
- Prokinetics: domperidone, erythromycin, metoclopramide
- N-acetylcysteine
- Lower GIT: malabsorption, bacterial overgrowth, diarrhoea, constipation, bloating, pain, anorectal dysfunction
- Nutritional supplements, advice from a dietician
- Creon
- Probiotics
- Cyclical antibiotics (for example, ciprofloxacin for ten days)
- Stool softeners
What is the outcome for patients with systemic sclerosis?
There is no cure for systemic sclerosis. Survival is determined by the disease subset and internal organ manifestations. Interstitial lung disease and pulmonary artery hypertension account for almost two-thirds of deaths related to systemic sclerosis.
Proactive and routine annual screening allows early intervention with disease-modifying drugs. These have led to an improvement in prognosis and long term outcomes in recent years.;