What is chilblain lupus erythematosus?
Chilblain lupus erythematosus (LE) is a rare variant of chronic cutaneous LE originally described by Jonathan Hutchinson in 1888 [1].
The term ‘chilblain’ is derived from Anglo-Saxon terms ‘chill’ referring to ‘cold’ and ‘blegen’, which is a synonym for ‘sore’.
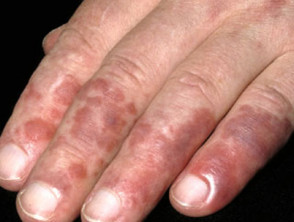
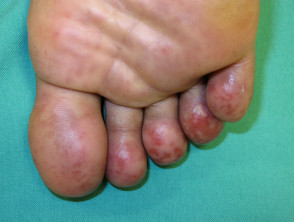
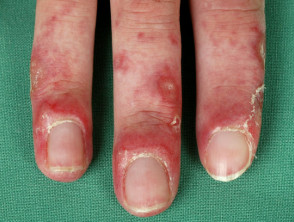
Lupus chilblains
Who gets chilblain lupus erythematosus?
Chilblain LE is an under-reported entity. In a 2008 review, the authors stated that 70 cases have been reported in the literature. It mainly occurs in adults but can occur at any age [2].
Chilblain LE occurs as both a sporadic and inherited condition. Sporadic cases are the most common and occur predominantly in middle-aged women [3], while familial chilblain LE presents in early childhood [4].
Associations between chilblain LE and anorexia nervosa, intestinal lymphoma, and pregnancy have been described in the literature.
What causes chilblain lupus erythematosus?
The pathogenesis of the sporadic form of chilblain LE remains unknown [2].
It is believed a cold stimulus provokes vasoconstriction or microvascular injury with occlusion of the capillary bed and diminished circulation, leading to hyperviscosity and stasis of the skin, which is exacerbated by reduced blood temperature [2,3].
These pathophysiological changes in the flow and microvasculature may be aggravated by immunological anomalies (eg, rheumatoid factor or autoantibodies).
In those with a family history or onset of chilblain LE in childhood, the condition may be a result of a mutation in the TREX1 or SAMHD1 genes [5].
- TREX1 is a gene located on chromosome 3 that encodes a nuclear protein with a role in DNA repair and proofreading for DNA polymerase.
- SAMHD1 is a gene located on chromosome 20 that encodes a host restriction nuclease with a role in the innate immune response.
These mutations in TREX1 and SAMHD1 are inherited in an autosomal-dominant manner [4,6]. Heterozygous mutation (mutation of one allele) in these genes lead to the accumulation of nucleic acids (eg, double-stranded DNA, RNA–DNA duplexes).
TREX1 mutations have also been described in other conditions related to microvascular endothelial dysfunction, such as dominant retinal vasculopathy and cerebral leukodystrophy.
What are the clinical features of chilblain lupus erythematosus?
Chilblain LE begins as red or dusky purple patches, papules, and plaques that are initiated or exacerbated by exposure to cold and moisture in a cool climate. The lesions are often pruritic and painful.
The common sites involved are the fingers, toes, heels, and soles of the feet. Less frequently, the lesions may be present on the nose, ears, palms, knuckles, elbows, knees, and lower legs.
Other features may include:
- Ulceration
- Hyperkeratotic fissuring
- Depigmentation similar to vitiligo
- Raynaud phenomenon.
People living in regions with warmer and drier climates may not develop typical chilblains.
In some circumstances, chilblains may represent lupus pernio with pre-existing LE rather than as chilblain LE [7]. Although the two entities are clinically similar and both are associated with systemic disease (sarcoidosis and systemic lupus erythematosus, respectively), the prognosis and treatment regimens are distinct [7].
What are the complications of chilblain lupus erythematosus?
Complications of chilblain LE include:
- Secondary bacterial skin infection
- Koebnerisation
- Progression to systemic lupus erythematosus (SLE).
Follow-up is advised for those with features of SLE (eg, malar rash).
How is chilblain lupus erythematosus diagnosed?
The Mayo Clinic diagnostic criteria for the diagnosis of chilblain LE comprises two major and four minor criteria [8]. To diagnose chilblain LE, both major criteria and at least one minor criterion need to be demonstrated.
Mayo Clinic diagnostic major criteria
The major criteria for chilblain LE are:
- Lesions in acral locations induced by exposure to cold or any decrease in temperature
- Evidence of LE in the skin lesions by histopathological examination or indirect immunofluorescence, including:
- A superficial and deep lymphocytic vascular reaction
- Fibrin deposition in reticular dermal blood vessels
- Interface dermatitis with degeneration at the dermoepidermal junction with an accumulation of immunoglobulin (Ig)M, IgA, and complement C3.
Mayo Clinic diagnostic minor criteria
The minor criteria for chilblain LE are:
- Coexistence of SLE or other forms of cutaneous LE
- Response to anti-LE therapy
- Negative results of cryoglobulin
- Negative results of cold agglutinin studies.
Chilblain LE is often associated with several immunological makers including:
- Anti-SSA/Ro antibodies
- Hypergammaglobulinaemia
- Rheumatoid factor
- Anti-nuclear antibodies
- Antiphospholipid antibodies
- Positive latex agglutination tests [2].
Raynaud phenomenon may coexist [2].
Lesions persisting beyond the colder months, positive antinuclear antibody tests, and concomitant features of the American College of Rheumatology criteria for SLE distinguish chilblain LE from idiopathic chilblains [9].
What is the differential diagnosis for chilblain lupus erythematosus?
Other skin conditions that can be confused with chilblain LE include perniosis (ordinary chilblains), acrocyanosis, Raynaud phenomenon, lupus pernio, livedo reticularis, dermatomyositis, cold panniculitis, and cold urticaria.
Perniosis
Perniosis (or usual chilblains) is characterised by:
- A sensation of itching, burning, or pain
- Symmetrical erythrocyanotic discolouration of acral skin
- Non-specific histology comprising dermal oedema with superficial and deep lymphohistiocytic infiltrate and peri-eccrine accentuation.
Acrocyanosis
Acrocyanosis is characterised by:
- Painless lesions in most cases
- Dusky red to purple discolouration
- Acral distribution.
Raynaud phenomenon
Raynaud phenomenon is characterised by:
- The sequence of well-demarcated pallor, followed by blue then red discolouration on rewarming
- Absence of ulceration (with the idiopathic form).
Livedo reticularis
Livedo reticularis is characterised by:
- Bluish, broad, reticulated patches
- An association with:
- Haematological disorders (eg, antiphospholipid syndrome)
- Cutaneous vasculitis (eg, polyarteritis nodosa)
- Infections (eg, syphilis)
- Medication (eg, minocycline).
Dermatomyositis
Dermatomyositis can cause nail fold erythema, telangiectasia, and nailfold inflammation similar to chilblain LE. Other clinical features of dermatomyositis include:
- Muscle weakness (myositis)
- Facial rash, including heliotrope (mauve) eyelids
- Photosensitivity
- Göttron papules (dry red plaques over the knuckles).
Cold panniculitis
Cold panniculitis is characterised by:
- Erythematous, indurated plaques that develop at sites of cold exposure
- Location on the cheeks and chin
- Occurrence in children and young women
- Resolution within 2 weeks.
Cold urticaria
Cold urticaria is a form of chronic inducible urticaria characterised by:
- Cold-induced weals and angioedema
- Systemic symptoms in some patients.
What is the treatment for chilblain lupus erythematosus?
General measures
Protection from the cold should be emphasised and may include:
- Avoidance of cold and moist environments where possible
- Ensuring that the home and workplace are well insulated and heated
- Wearing gloves, thick woollen socks, and comfortable protective footwear
- Soaking hands in warm water to warm them up throughout the day
- Engaging in physical activity to maintain central body temperature.
Smoking cessation should also be encouraged, due to the vasoconstrictive effects of nicotine.
Medications
Medication may be necessary for patients with recalcitrant lesions and for secondary bacterial skin infections. A therapeutic approach has been proposed based on the level of evidence of the therapies in chilblain LE [2].
Medicines used include the following:
- Antibiotics (for secondary bacterial skin infections)
- Topical steroids
- Systemic calcium channel blockers
- Systemic steroids
- Mycophenolate mofetil
- Antimalarial agents (eg, hydroxychloroquine)
- Tacrolimus.
Surgery
Patients with chilblain LE have successfully been treated in certain circumstances by excision and repair using a full-thickness graft derived from an unaffected area.
What is the outcome for chilblain lupus erythematosus?
The majority of patients respond well to symptomatic treatment.
In those with the sporadic form of chilblain LE, 18% of individuals will eventually develop SLE; there is no evidence of progression to SLE in cases of familial chilblain LE.